MHDMF: Prediction of miRNA–disease associations based on Deep Matrix Factorization with Multi-source Graph Convolutional Network

摘要
越来越多的研究证明,微小 RNA (miRNA)是影响各种疾病的不同生物过程中的重要生物标志物。作为对高成本湿实验方法的有力补充,大量的计算预测方法应运而生。然而,在有效利用高度假阴性关联和多来源信息寻找潜在关联方面仍然存在挑战。在这项工作中,我们开发了一个端到端的计算框架,称为 MHDMF,它整合了异质网络上的多源信息,发现潜伏的疾病与 miRNA 之间的关联。由于 miRNA 疾病关联中存在高度假阴性,MHDMF 利用多源图卷积网络(GCN)通过重新制定 miRNA 疾病关联评分矩阵来纠正假阴性关联。评分矩阵的重新制定是基于miRNA,基因和疾病之间的不同的相似性谱和已知的关联。然后,基于重新制定的 miRNA-疾病关联评分矩阵,MHDMF 使用深度矩阵分解(Deep-basel,DMF)来预测 miRNA-疾病关联。实验结果表明,该框架在 miRNA-疾病关联预测任务上优于高度相关的比较方法。此外,个案研究表明,MHDMF 可能是一种方便有效的工具,并可能为 miRNA 疾病的关联预测提供一个新的思路。
关键词:MiRNA–disease associations,Multi-source information,Graph convolutional networks,Deep matrix factorization,End-to-end framework。

目录

Disease gaussian interaction profile kernel similarity
miRNA gaussian interaction profile kernel similarity
2.5.miRNA-疾病关联的关联评分矩阵重构(The association score matrix reformulation of miRNA–disease association)
Multi-source information learning with GCN
Multi-source information importance learning with attention mechanism
3.3.1. Performance of MHDMF and its variants
3.3.2. Performance of MHDMF on different heterogeneous networks
3.4. Parameter analysis of MHDMF
3.4.3. Dimensionality of CNN and DNN features
3.4.5. Dimensionality of DMF feature
1.引言
miRNA (microRNA)是一种短的单链内源性非编码 RNA (ncRNA)分子,长约21-23个核苷酸。越来越多的证据表明,miRNA 在细胞生长、增殖、分化等多种生物过程中起着至关重要的作用[1-3]。它们的功能障碍密切影响各种疾病的发生和发展[4-6]。此外,2019冠状病毒疾病正在破坏世界。Yu 等[7]发现透明质酸的增加可以作为2019冠状病毒疾病发展为严重疾病趋势的关键指标。从2019冠状病毒疾病发病的关键分子机制入手,他们发现细胞核内的 miRNA 可以通过靶向增强子来激活基因表达,从而导致透明质酸的积累。抑制细胞核中的 miRNA 可以减少透明质酸的积累,这可能是阻止2019冠状病毒疾病临床进展的有效策略。识别潜在的疾病相关的 miRNA 将有助于我们进一步了解其发病机制并进行精确的治疗。然而,传统的生物实验不仅耗费时间和金钱,而且效率低下,容易受到外部世界的影响。基于未经充分证实的相关性,仍然很难清楚地了解 miRNA 在疾病中的功能和作用[8-10]。随着技术的发展,多源信息的快速积累为开发高性能和低成本的计算方法创造了巨大的机会[11-14]。因此,作为湿实验方法的一个很好的补充,许多计算预测方法已经涌现出来用于进一步的验证实验[8-10,15]。将现有的流行计算方法分为两类: 基于网络的和基于机器学习的[16]。
基于网络的方法是基于一个核心假设,即与相似疾病相关的 miRNA 往往具有相似的功能,反之亦然[8-10]。Jiang等人将 miRNA-疾病关联预测真实化为链接预测。他们提出了第一个计算模型,它使用一个离散的超几何概率分布,在关联网络上提供直接的边缘网络信息。对于全局网络信息,一些方法是基于重新启动算法的随机游走[18-22]。[23,24]提出了基于路径的方法来预测 miRNA 与疾病的关联。Yu 等人首先构建一个 miRNA 疾病异质网路,根据全球线性邻域对 miRNA 疾病对进行排序。Chen 等人分别探讨了全球网络相似性以捕获疾病与 miRNA 之间的相似关系[26,27]。Chen 等人通过构建二分网络投影模型来整合它们的相似性,从而推断 miRNA 与疾病的关联[28]。尽管这些基于网络的方法性能良好并且有很好的解释,但是性能受到网络中已知链接数量的严重影响。
基于机器学习的方法利用 miRNA 和疾病的生物学特性来训练潜在 miRNA-疾病关联预测模型。自然语言处理(NLP)作为一种机器学习方法,是生物医学文本挖掘领域中描述生物分子间关联的重要方法。Abdulkadhar 等人基于多尺度 Laplacian 图核结合词汇句法模式提取具有参数的生物事件[29]。矩阵分解(MF)方法已成功应用于鉴定疾病相关生物标志物。Chen 等人提出了一种基于矩阵分解的 MDHGI 方法来推断 miRNA 与疾病的关联。Fu 等人使用 MF 来探索和开发异构数据源的内在和共享结构[31]。然而,MF 仅提取线性特征,不足以模拟 miRNA 与疾病之间复杂的关联。近年来,深度学习也被广泛应用于解决 miRNA-疾病关联预测问题。Xuan等人提出了两种基于双卷积神经网络的模型来鉴定与疾病相关的 miRNA。曾等人使用结合 SVD 和 DMF 的端到端模型来提取线性和非线性特征来推断潜在关联[34]。尽管如此,他们中的大多数忽略了上下文信息。Peng 等人构建了一个 miRNA 基因疾病三层异质网路,并设计了一个结合基于网络的方法和深度学习的 MDACNN 框架,用于 miRNA 疾病关联鉴定。MDA-CNN 使用基于网络的方法 CIPHER [36]来获取关于异质网路的上下文信息。基于图的方法可以利用上下文信息使模型具有良好的性能。Han 等人提出了一个结合 GCN 和 MF 的框架来捕获非线性相互作用并利用测量的相似性[37]。Ding 等人利用变异图形自动编码器预测 miRNA 与疾病的关联[38]。总之,所有这些方法有助于更有效和更好地理解 miRNA 在疾病中的功能和作用。尽管已经作出了巨大的努力来探索潜在的 miRNA 疾病关联,但在 miRNA 疾病关联预测中仍然存在一些挑战,例如高假阴性关联和多源信息利用不足[8,39,40]。首先,许多计算方法强烈依赖于通过湿实验测试的 miRNA-疾病关联。不幸的是,在 miRNA 疾病数据库中存在许多假阴性关联。数据库通常使用1和0来表示 miRNA 与疾病之间的关联,其中1表示关联,0表示未确定的关联,而不是没有关联。仍然有很少的关联是由湿实验验证,导致数据的不平衡。数据中大量的“0”并不意味着 miRNA 真的与疾病无关。因此,高的假阴性将影响该方法的性能和可解释性。其次,方法可以通过集成多种多源信息来提高性能。以往的许多研究通常采用简单的平均或线性加权策略来整合 miRNA 与疾病的相似性特征。他们忽略了不同的信息对同一个预测任务的贡献可能不同。此外,他们也有多阶段和依赖于手工制作的中间结果,可能会影响结果。值得注意的是,大多数方法都集中在两个感兴趣的特定实体(例如,miRNA 和疾病)上,这些方法不适用于考虑具有两种以上实体的网络。减少假阴性,充分利用多源信息,区分不同信息来源的重要性,可以提高 miRNA-疾病关联预测性能。然而,目前还没有完整的端到端框架来应对这些挑战。
在这项工作中,为了克服上述挑战,我们开发了一个新的端到端的多源异质网路信息集成框架与深度矩阵分解(MHDMF) ,用于预测潜在的 miRNA-疾病关联。端端框架 MHDMF 具有以下优点:
1.我们提出了一个新的框架 MHDMF 首先使用 GCN 基于三层异质网路在 miRNA 和疾病之间产生嵌入。这三层异质网路包括 miRNA、基因和疾病,如图1所示。MHDMF 由三个模块组成: (i)多源异质网路构建、(ii)关联评分矩阵重构和(iii)潜在关联预测。
2.MHDMF 首先将 GCN 和 DMF 有效地结合成一个端-端框架,用于预测任务中的潜在关联。该框架通过重新构建基于多源 GCN 的 miRNA-疾病关联评分矩阵来纠正假阴性关联。它进一步使用 DMF 对重新制定的评分矩阵考虑显性和隐性反馈,以预测潜在的 miRNA 疾病的关联。
3.为了评估 MHDMF 的能力,我们将其与五种最先进的方法在5折交叉验证(5CV)和一次性交叉验证(LOOCV)下进行了比较,MHDMF 获得了最佳的结果。此外,我们还进行了烧蚀实验,验证了 MHDMF 各部分的有效性,并进行了参数分析,说明了参数的选择。最后,对淋巴瘤、结肠癌和神经胶质瘤进行病例研究。实验结果表明,MHDMF 是一种方便、有效的高成本临床生物学实验工具。
2. 材料和方法
2.1. miRNA- 基因-疾病异质信息网络
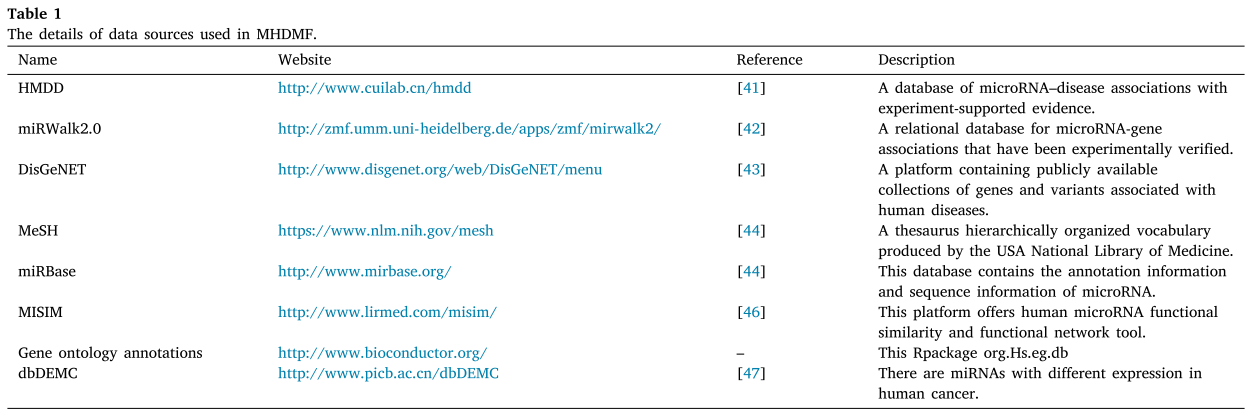
这组正面的 miRNA 疾病相关性可以从人类微 RNA 疾病数据库(HMDD)中下载[41]。HMDD 是一个手动收集的数据库,用于人类 miRNA 疾病与实验证实的证据的关联。实验验证的 miRNA 基因关联从全面的存档 miRWalk2.0数据库下载[42]。已知的疾病-基因关联从 DisGeNET [43]下载。我们只是手工管理疾病-基因关联,并交叉数据集以去除与疾病和 miRNA 无关的基因。同时,我们还从美国国家医学图书馆(MeSH)[44]下载了疾病的语义树。MiRNA 序列信息从1917年人类 miRNA 的 miRBase Release22[45]中下载。我们筛选出在 MeSH 描述符或 miRBase 记录中没有相应名称的 miRNA-疾病关联。
我们整合多源信息来建立整个 miRNA 疾病异质性信息网络,如图1所示。这些异质网路包括 miRNA 的功能、序列和 GIP 核相似性网络、 疾病的Jaccard、语义和 GIP 核相似性网络,以及实验上有效的 miRNA-疾病、 miRNA-基因、疾病-基因关联。在这项工作中,所有miRNA 和疾病的相似性网络被视为图的边缘加权。将 miRNA 基因和疾病基因的关联矩阵分别作为 miRNA 和疾病的边缘加权图的特征。

2.2.模型框架
我们开发了一个名为 MHDMF 的端到端框架用于潜在的 miRNA 疾病关联预测。事实上,各种异构数据包含不同的表示或生物信息学的互补信息[48-50]。此外,已经证明,整合不同的信息或利用互补的信息可以使模型具有更好的性能[51,52]。此外,通过湿实验验证的 miRNA-疾病关联是非常不充分的,并且存在许多假阴性关联[39]。因此,我们通过整合不同的相似性谱和相互作用谱(表1)来构建 MHDMF,以重新制定潜在关联推断任务的 miRNA-疾病关联评分矩阵。
如图2所示,MHDMF 是一个用于多源信息集成的端到端深度学习框架,由三个模块组成: (i)多源异质网路结构(图1) ,(ii)关联评分矩阵重构(图2I)和(iii)潜在关联预测(图2II)。在多源信息整合部分,我们构建了一个三层异质网路,包括 miRNA、基因和疾病的相似性谱和相互作用谱。在关联评分矩阵重构部分,我们首先使用 GCN 编码 miRNA、疾病和基因的不同相似性和相互作用特征。然后利用注意机制来了解不同信息源的重要性。结合注意机制的研究结果,我们可以获得 miRNA 与疾病的特征嵌入。 最后,将特征嵌入与小 RNA 和疾病的 DNN 嵌入融合,得到最终的嵌入结果。这些最终的嵌入被用于 MF 重新制定 miRNA-疾病关联评分矩阵。在潜在关联预测部分,采用 DMF 进行预测任务。MHDMF 使用 DMF 直接从 miRNA 与疾病的关联中提取 miRNA 和疾病的最终特征。DMF 扩展了非线性表示的基本潜因子模型,而不是线性表示。

 2.3.疾病相似性度量
2.3.疾病相似性度量
Disease Jaccard similarity
为了避免依赖于 miRNA 和疾病之间现有的关联,使用疾病和基因之间的关联可以计算疾病 Jaccard 相似性。Jaccard 相似性(系数)是用来比较样本集的相似性和多样性的统计量。Jaccard 相似度可以度量有限样本集的相似度,定义为两个集合的交集大小和并集大小之间的比例[53]。这项工作中的疾病-疾病关联(DDA)是通过计算从 DisGeNET 数据库中获得的疾病对之间的共享基因数来获得的。我们通过每个疾病对的 Jaccard 相似性 DSJ (di,dj)评估疾病之间共享基因的比例,Jaccard 相似性的定义可以定义如下:

其中![]() 是疾病
是疾病![]() 相关的基因集。
相关的基因集。
Disease semantic similarity
近年来,Wang 等[54]提出的术语相似度计算方法在疾病的语义相似度计算中得到了广泛的应用[3,8,9,39]。根据 meSH 疾病描述符,疾病可以由相应的有向无环图结构(DAG)表示,节点代表疾病,每个定向边代表一般疾病术语到特定疾病术语。越是相似的疾病,它们共享的 DAGs 中越是常见的部分。因此,我们用 Eq(2). 方法计算了疾病 di 和疾病 dj 之间的疾病语义相似度 DS1S (di,dj)。


一种疾病出现的 DAGs 越多,它就越常见。这种疾病出现在较少的 DAGs 中,表明这种疾病是独特的。根据语义相似度计算,同一层次上不同祖先疾病的语义贡献值是相同的。然而,对于独特的疾病,其语义贡献值应区别于常见疾病的语义贡献值。受前人工作[55]的启发,我们采用第二种语义相似性方法来区分独特疾病和常见疾病的语义贡献。相应地,疾病 di 和 dj 之间的第二疾病语义相似度 DS1S (di,dj)的计算可以写成如下:


根据以前的工作[56] ,我们合理地将上述两种语义相似性计算的结果合并为疾病 di 和 dj 之间的疾病语义相似性 DSS (di,dj) ,其可以描述如下:
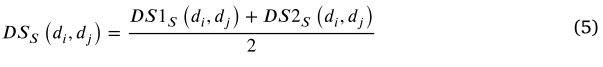
Disease gaussian interaction profile kernel similarity
高斯相互作用分布(GIP)核函数是一个径向标量对称函数。它可以捕获生物实体对(如疾病和 miRNA)相互作用网络的拓扑特征[57]。这是一个很好的度量样本之间的相似性,相似的样本可以更好地聚类在描述相似性的空间。因此,GIP 核相似性经常用于计算生物实体之间,包括疾病和 miRNA [58,59]。疾病之间的疾病 DSG (di,dj)的 GIP 核相似性可以定义如下:
![]()
其中 Adj (di)和 Adj (dj)是疾病 di 和 dj 的二元向量,分别对应关联邻接矩阵中的 ith 和 jth 列。![]() 是控制核带宽的调节参数。
是控制核带宽的调节参数。

2.4. miRNA相似性度量
miRNA functional similarity
我们可以根据 Wang 等人提供的 miRNA 功能相似性计算推导 miRNA 功能相似性[60] ,这是基于表型相似的疾病更可能与功能相似的 miRNA 相关的核心假设,反之亦然。我们从 MISIM 数据库下载功能相似性得分矩阵[46]。然后通过构建 MSF 矩阵来存储 miRNA 的功能相似性,其中 MSF (mii,mij)表示 miRNA mii 和 mij 之间的功能相似性得分。
miRNA sequence similarity
我们可以基于 Needleman-Wunsch 算法[61]计算 miRNA 序列相似性矩阵 MSS。MiRNA 的序列信息从 miRBase [44]下载,并且使用 R 包 Biostring 中的成对比对函数来定量相似性评分。然后,矩阵由序列比对分数 Mseq (mii,mij)构成,匹配值和失配值分别设置为1和 -1[62]。为了确保全局一致性,序列比对得分矩阵需要归一化到如下范围[0,1] :


miRNA gaussian interaction profile kernel similarity
同样,miRNA mii 和 mij 之间的 miRNA MSGIP (mii,mij)的 GIP 核相似性可以通过以下公式获得:
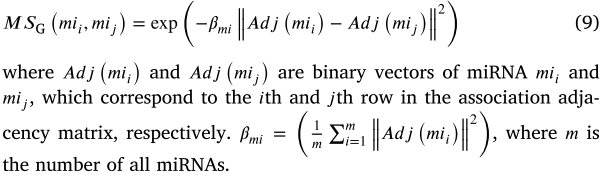
2.5.miRNA-疾病关联的关联评分矩阵重构(The association score matrix reformulation of miRNA–disease association)
这一部分有三个组成部分,包括(1)用于 miRNA-疾病关联评分矩阵重新制定的多源 GCN,(2)用于多源数据重要性学习的注意机制,(3)用于最终重新制定的 CNN,DNN 和 MF。
Multi-source information learning with GCN
某些以前的工作[63,64]表明,GCN 及其变体有利于从网络学习。它们保留了网络拓扑的节点级向量嵌入,提高了基于网络的链路预测性能。在 MHDMF,我们使用 GCN 进行特定源的学习,对每个源数据训练一个 GCN 来实现关联分数矩阵重构组件。
如图2a 所示,每个 GCN 的作用是学习多源数据在边加权图![]() 上的有用嵌入。因此,GCN 模型有两个输入。一个输入是一个表示图结构描述的邻接矩阵
上的有用嵌入。因此,GCN 模型有两个输入。一个输入是一个表示图结构描述的邻接矩阵 ![]() ,其中 a 是节点数量。另一个输入是由节点特征构成的矩阵
,其中 a 是节点数量。另一个输入是由节点特征构成的矩阵![]() ,其中
,其中 ![]() 是特征的维数。
是特征的维数。
在这项工作中,原始的 ![]() 是 m miRNA 或 n 种疾病的相似矩阵,这是一个边加权图。原始 F 是 miRNA-基因 或 疾病-基因 的归一化关联矩阵。对于miRNA,一个 GCN 可以通过叠加以下多个卷积层来构建:
是 m miRNA 或 n 种疾病的相似矩阵,这是一个边加权图。原始 F 是 miRNA-基因 或 疾病-基因 的归一化关联矩阵。对于miRNA,一个 GCN 可以通过叠加以下多个卷积层来构建:

其中 ![]() 为输入,
为输入,![]() 为第 l GCN 层的权矩阵,两者均在 vth miRNA 相似性边权图上。
为第 l GCN 层的权矩阵,两者均在 vth miRNA 相似性边权图上。![]() 表示非线性激活函数。为了更有效地训练广义连接网络,进一步修改了邻接矩阵
表示非线性激活函数。为了更有效地训练广义连接网络,进一步修改了邻接矩阵 ![]() 。然后,我们可以得到嵌入的 miRNA 节点如下:
。然后,我们可以得到嵌入的 miRNA 节点如下:

其中 ![]() 是
是 ![]() 的对角节点度矩阵。
的对角节点度矩阵。![]() 是一个保持节点本身重要性的自循环,而
是一个保持节点本身重要性的自循环,而 ![]() 是一个单位矩阵。同样地,我们可以通过以下方法得到嵌入在第 t 个疾病相似性边权图上的疾病节点:
是一个单位矩阵。同样地,我们可以通过以下方法得到嵌入在第 t 个疾病相似性边权图上的疾病节点:

疾病邻接矩阵的维数是 ![]() 。最后,我们可以通过多层的 GCN 从多个来源的数据中得到 miRNA 和疾病的嵌入矩阵。
。最后,我们可以通过多层的 GCN 从多个来源的数据中得到 miRNA 和疾病的嵌入矩阵。
Multi-source information importance learning with attention mechanism
我们将 miRNA-疾病联系预测作为一项推荐任务。在这样的任务中,由于从一个视角嵌入的性能可能达不到推荐任务的期望,许多作品试图融合更多的视角来获得嵌入。然而,不同的视角对嵌入学习应该有不同的贡献[65]。类似地,我们使用多通道注意机制来学习多源信息的重要性。
从图(2b) ,我们将上 GCN 的嵌入矩阵叠加为一个嵌入张量。我们把嵌入矩阵看作是注意层的通道。通过建立每个信道的重要性模型,可以增强或抑制不同信道对多源信息重要性的学习。
具体来说,对于疾病,我们需要通过全局平均池化将每种疾病嵌入张量中的矩阵压缩到一个真正的 ![]() ,如下所示:
,如下所示:

其中 ![]() 是疾病的 cth 嵌入矩阵。将疾病嵌入
是疾病的 cth 嵌入矩阵。将疾病嵌入 ![]() 挤压生成
挤压生成 ![]() ,
,![]() 为通道数。通道的重要性通过注意力机制计算为注意力权重:
为通道数。通道的重要性通过注意力机制计算为注意力权重:
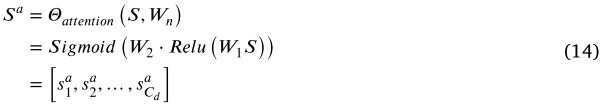
其中 ![]() 是训练参数。
是训练参数。
最后,我们得到了具有注意力权重的归一化通道嵌入:
![]()
如前所述,我们可以得到疾病的通道信息 ![]()
![]() ,以及 miRNA
,以及 miRNA ![]() 。
。
Final reformulation
在增强通道嵌入的基础上,利用CNN生成 miRNA 和疾病的特征嵌入。我们通过两个全连接层分别从原始的 miRNA-疾病关联中获得 miRNA 和疾病的 DNN 嵌入(如图2c 所示)。通过上述步骤,如图2d 所示,我们可以通过结合特征嵌入和 DNN 嵌入来获得 miRNA ![]() 和疾病
和疾病 ![]() 的最终嵌入。我们利用 MF 重构了 miRNA-疾病关联评分矩阵
的最终嵌入。我们利用 MF 重构了 miRNA-疾病关联评分矩阵 ![]() ,它可以表示为:
,它可以表示为:
![]()
2.6.通过深度矩阵分解预测 miRNA 与疾病的关联(The prediction of miRNA-disease association by deep matrix factorization)
MF 在基于潜在因素模型的推荐任务中表现出很强的有用信息挖掘能力[66]。它已成功地应用于鉴定生物实体[31,67,68]之间的潜在联系,包括疾病,药物,基因,miRNA,lncRNA 等。对 MF 预测矩阵的得分进行排序的显性知识已经在早期的潜在关联预测方法中得到了深入的应用[69]。为了克服生物实体之间关联的稀疏性,额外的信息被整合到 MF 中,例如具有 miRNA 和疾病相似网络的相似性约束矩阵分解[70]。然而,仅对观察到的关联进行建模不足以提出良好的前 N 关联推荐[71]。Lu 和 Tsang 等人利用隐式反馈对关联矩阵进行因子分解[34,72]。此外,只有 MF 提取的线性特征不足以模拟 miRNA 与疾病之间如此复杂的关联。DMF 可以通过显性和隐性反馈从 miRNA-疾病关联矩阵中提取非线性特征,提高预测性能。
图2II 显示了三个步骤。首先,基于重新构建的 miRNA-疾病关联评分矩阵 ReM,提取行和列向量分别表示 miRNA 和疾病的原始特征。MiRNA ![]() 和疾病
和疾病 ![]() 的关联模式包含在每个行向量
的关联模式包含在每个行向量 ![]() 和列向量
和列向量 ![]() 中。
中。![]() 被认为是 miRNA 与所有疾病之间的关联,而
被认为是 miRNA 与所有疾病之间的关联,而 ![]() 被认为是 jth 疾病与所有 miRNA 之间的关联。在原始的 miRNA-疾病关联矩阵 M 中,1表示与实验支持的已知关系(显性反馈) ,而0表示未知关系而不是没有关系(隐性反馈) ,因此具有较高的假阴性。仅仅使用显式反馈不足以获得良好的性能。为了减少假阴性,在 ReM 中,我们预测一些未知关联的得分。我们保留了 miRNA 与疾病之间的原始关联,其值为1。隐式反馈由非一个值表示,而不是仅由0表示。这样,我们可以通过进一步利用关联模式组成的隐式反馈来提高性能。其次,
被认为是 jth 疾病与所有 miRNA 之间的关联。在原始的 miRNA-疾病关联矩阵 M 中,1表示与实验支持的已知关系(显性反馈) ,而0表示未知关系而不是没有关系(隐性反馈) ,因此具有较高的假阴性。仅仅使用显式反馈不足以获得良好的性能。为了减少假阴性,在 ReM 中,我们预测一些未知关联的得分。我们保留了 miRNA 与疾病之间的原始关联,其值为1。隐式反馈由非一个值表示,而不是仅由0表示。这样,我们可以通过进一步利用关联模式组成的隐式反馈来提高性能。其次,![]() 和
和 ![]() 被视为输入,它们被输入到两个全连接层中,将 miRNA 和疾病投射到潜在的结构化空间中。过程如下:
被视为输入,它们被输入到两个全连接层中,将 miRNA 和疾病投射到潜在的结构化空间中。过程如下:

其中 ![]() 是
是 ![]() 中间隐藏层,
中间隐藏层,![]() 是隐藏层的个数。
是隐藏层的个数。![]() 和
和 ![]() 分别是
分别是![]() 隐层上的权矩阵和偏置项。
隐层上的权矩阵和偏置项。![]() 是一个非线性激活函数,我们在这里使用Rectified Linear Unit(ReLU)。
是一个非线性激活函数,我们在这里使用Rectified Linear Unit(ReLU)。
在这项工作中,有两个多层网络提取 miRNA 和疾病的特征分别如下:

其中 ![]() 分别代表第一层 miRNA 和疾病的加权矩阵。
分别代表第一层 miRNA 和疾病的加权矩阵。
第三,得到了 miRNA 和疾病的最终特征矩阵 ![]() 和
和 ![]() 。通过 MF 可以得到最终的 miRNA-疾病关联预测矩阵
。通过 MF 可以得到最终的 miRNA-疾病关联预测矩阵 ![]() ,其结果如下:
,其结果如下:
![]()
对于 M 中的值,![]() 越高,miRNA i 和疾病 j 之间的关联就越可能,反之亦然。
越高,miRNA i 和疾病 j 之间的关联就越可能,反之亦然。
将预测矩阵 M 与原始矩阵 A 之差的 Frobenius 范数最小化,得到预测矩阵 M 与原始矩阵 A 之间的均方误差作为损失函数。损失函数显示如下:
![]()
3.Experiments
3.1.Experiment settings
在实验中,利用5CV 和 LOOCV 来评估 MHDMF 对 miRNA 与疾病之间潜在关联的预测性能。在{2,3,4,5}中选择 GCN 层数 l,在{1,2,3}中选择 DNN 层数,在{48,64,96,128}中选择 CNN 和 DNN 维度,在{1,2,3}中选择 DMF 层数![]() ,在{32,48,64,96}中选择 DMF 特征维度,在{0.1,0.01,0.001}中选择学习率。我们选择 ReLU 作为一个激活函数,并在这个框架中使用 Adam 优化器。
,在{32,48,64,96}中选择 DMF 特征维度,在{0.1,0.01,0.001}中选择学习率。我们选择 ReLU 作为一个激活函数,并在这个框架中使用 Adam 优化器。
对 MHDMF 框架的实验分析基于 HMDD 和 DisGeNET 数据集。我们比较 MHDMF 和比较方法来进一步评估性能。评估指标包括 ROC曲线曲线下的面积(AUC)、精确/召回曲线下的面积(AUPRC)、 F1分数、Recall和Precision。此外,通过计算方法获得的高排名的疾病相关 miRNA 有助于进一步分析生物学实验。
3.2.Comparison with highly related methods
证明了 MHDMF 方法的有效性,并与近年来提出的其他先进方法进行了比较。在这项工作中,我们使用5CV 和 LOOCV 来评估 MHDMF 和其他方法对同一 miRNA 疾病基因数据集的有效性。比较方法中的参数设置与项目中的参数设置相同。
3.2.1.Baselines
MDA-SKF [73]提出相似核融合(SKF)分别融合 miRNA 和疾病的多个相似性网络。然后,拉普拉斯正则化最小二乘法被用来揭示潜在的 miRNA 疾病的关联。
基于包含其相似性概况的 CIPHER 方法,基于 miRNA 基因疾病异质网路,MDA-CNN [35]捕获了 miRNA 疾病的相互作用特征。自动编码器的特征降维,然后,CNN 被用来预测最终的联系。
NIMCGCN [10]利用 GCN 在两个相似性网络上分别提取 miRNA 和疾病的特征。同时,将神经归纳矩阵补全作为预测模块。整个框架以端到端模式完成预测。
MMGCN [8]利用 GCN 和注意力机制,通过融合不同的相似性图谱分别产生 miRNA 和疾病的多视图嵌入,以便在 miRNA 和疾病发现之间建立可能的联系。
DMFCDA [72]使用三层 DMF,仅根据 miRNA 和疾病之间的原始关系推断潜在的 miRNA-疾病关联。整个框架以端到端的模式完成预测。
DMFMSF [40]通过 SKF 整合了两种类型的 RNA 和疾病的几个相似性谱,以产生特征,这些特征被用于通过加权 k 最近已知邻居(WKNKN)重新制定 RNA-疾病关联邻接矩阵。基于重新构建的邻接矩阵,三层 DMF 和奇异值分解,分别提取 RNA 和疾病的非线性和线性特征,并结合这些特征来预测 RNA 与疾病之间的关联标签。
3.2.2.性能比较
表2和表3给出了所有的比较结果,表明了 MHDMF 的合理设计。在这些高度相关的方法中,所提出的框架 MHDMF 在所有评价指标下都具有优越的性能。与多阶段框架(MDA-CNN 和 DMFMSF)相比,我们的框架是一个端到端的框架。它可以避免特征提取的不准确性和中间结果引起的误差累积。与基于 GCN 的方法(NIMCGCN 和 MMGCN)相比,MHDMF 使用 miRNA-gene-disease 相似性权重异质网路来学习更多的信息。此外,还对 GCN 模块的结果进行了进一步的分析,以更加合理地得到最终的结果。与基于 DMF 的方法(DMFCDA 和 DMFMSF)相比,MHDMF 在一定程度上减少了输入 DMF 的数据的假阴性。它综合了多源信息,重新构造了关联评分矩阵,使该框架具有更强的性能。总之,MHDMF 在潜在 miRNA-疾病相关性预测中是有效的,上述实验结果证实了这一点。

