今天小编和大家分享一个在KEGG通路分析中可能会经常用到的一个R包,名字叫Pathview。我将分五期进行,从快速上手,再到高级用法,最后在用实例对这个包做一个较全面的讲解。下面我们先简单了解一下这个包并学会快速使用它吧。
1. 简单介绍
Pathview包是最早在2013年发布的一个生信工具,该包的主要功能是基于基因通路的数据整合和可视化。它可以将用户的数据映射到特定的通路图上并显示出来。用户只需要提供自定义的数据并指定目标通路,Pathview就可以自动下载相应的kegg路径图数据,解析数据文件,将用户数据映射和整合到通路上,并显示出通路图。虽然Pathview是作为一个独立的R包 ,但它可以与其它通路和功能分析工具无缝集成,用于大规模和完全自动化的分析。
Pathview包是一个独立的生信分析R包,主要功能是基于基因通路的数据整合和可视化。该包可以被分为四个功能模块:
Downloader,下载KEGG通路图
Parser, 解析数据
Mapper, 将基因数据映射到通路图上
Viewer, 浏览结果
此外Pathway在数据整合方面有很强大的功能,包括:
它适用于基本上所有类型的映射到基因通路上的生物学数据
超过10种类型的基因或蛋白质ID,以及20种类型的化合物或代谢物ID
约4800个物种的路径以及KEGG词条
多种数据属性和格式,即连续/离散数据、矩阵/矢量、单个/多个样本数据等
2. 下载和安装
Pathview包是发布在BioConductior上的一个包,所以我们按照相应的方法安装即可。现在在BioConductor上可以下载到的是1.34.0版本
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("pathview")3. 快速上手
Pathview包中的主函数是pathview(),有着各种参数,使我们用到最多的函数。
Pathview在通路图数据的可视化时有两种输出格式,分别是原始的KEGG视图和Graphviz视图。前者将用户的数据呈现在原始的KEGG路径图上,因此很自然,更容易阅读。后者使用Graphviz引擎布置路径图;因此可以更好地控制节点或边的属性和路径拓扑结构。在使用时可以根据实际需要选择合适的输出格式。
我们下面举例会使用微阵列数据作为基因数据的例子,但Pathview同样适用于RNA-Seq数据和其他类型的基因/蛋白质的高通量数据。
3.1 加载基因数据
我们先以一个实例芯片数据为例,来快速上手Pathview。开始先加载我们的R包和一个包里自带的数据
library(pathview)
data("gse16873.d")数据是6个乳腺癌样本的基因芯片数据,列名为基因的ENTREZ ID。例子中的芯片数据是经过log2转化过的。
> head(gse16873.d)
DCIS_1 DCIS_2 DCIS_3 DCIS_4 DCIS_5 DCIS_6
10000 -0.30764480 -0.14722769 -0.023784808 -0.07056193 -0.001323087 -0.15026813
10001 0.41586805 -0.33477259 -0.513136907 -0.16653712 0.111122223 0.13400734
10002 0.19854925 0.03789588 0.341865341 -0.08527420 0.767559264 0.15828609
10003 -0.23155297 -0.09659311 -0.104727283 -0.04801404 -0.208056443 0.03344448
100048912 -0.04490724 -0.05203146 0.036390376 0.04807823 0.027205816 0.05444739
10004 -0.08756237 -0.05027725 0.001821133 0.03023835 0.008034394 -0.068607493.2 加载通路数据
下面加载一个包内自带的通路数据
data("demo.paths")观察一下通路数据的结构,是长度为5的列表。包含了通路的ID以及画图的一些参数数据。当然,我们在实际使用时没有必要将通路数据和画图参数做成一个列表,只需要在主函数内设置相应参数就好了。
> str(demo.paths)
List of 5
$ sel.paths: chr [1:3] "04110" "00620" "00640"
$ kpos1 : chr [1:3] "topright" "bottomright" "topright"
$ kpos2 : chr [1:3] "topright" "topright" "bottomright"
$ spos : chr [1:3] "bottomleft" "bottomleft" "topright"
$ offs : num [1:3] -1 -1 -0.83.3 使用pathview函数画通路图
我们先来看一下单个样本的基因表达量在“Cell Cycle”这条通路上的变化情况,只需要明确gene.data 和 pathway.id 两个参数即可。我们选择第一个样本的数据做展示,即DISC_1。一些细节如下:
pathview()函数是该包中的核心函数,拥有众多参数来调节通路图的细节
我们现在只提供基因数据和通路id,画一个最基本的形式
#KEGG view: gene data only
i <- 1
pv.out <- pathview(gene.data = gse16873.d[, 1], #取第一个样本
pathway.id = demo.paths$sel.paths[i], #选择第一个通路的id
species = "hsa", #人类样本
out.suffix = "gse16873", #输出文件的后缀
kegg.native = TRUE) #输出的图片形式为kegg画好后图片会被保存在当前的工作目录,我们可以找到并打开画好的图像
#展示出工作目录中的结果
> list.files(pattern="hsa04110", full.names=T)
[1] "./hsa04110.gse16873.png" "./hsa04110.png" "./hsa04110.xml"图像如下,可以看出:
这是我们看到的经典的KEGG通路图
该样本在该通路上基因的表达量映射在了相应基因处,此时展示的是基因的KEGG名称,而不是常规的Symbol
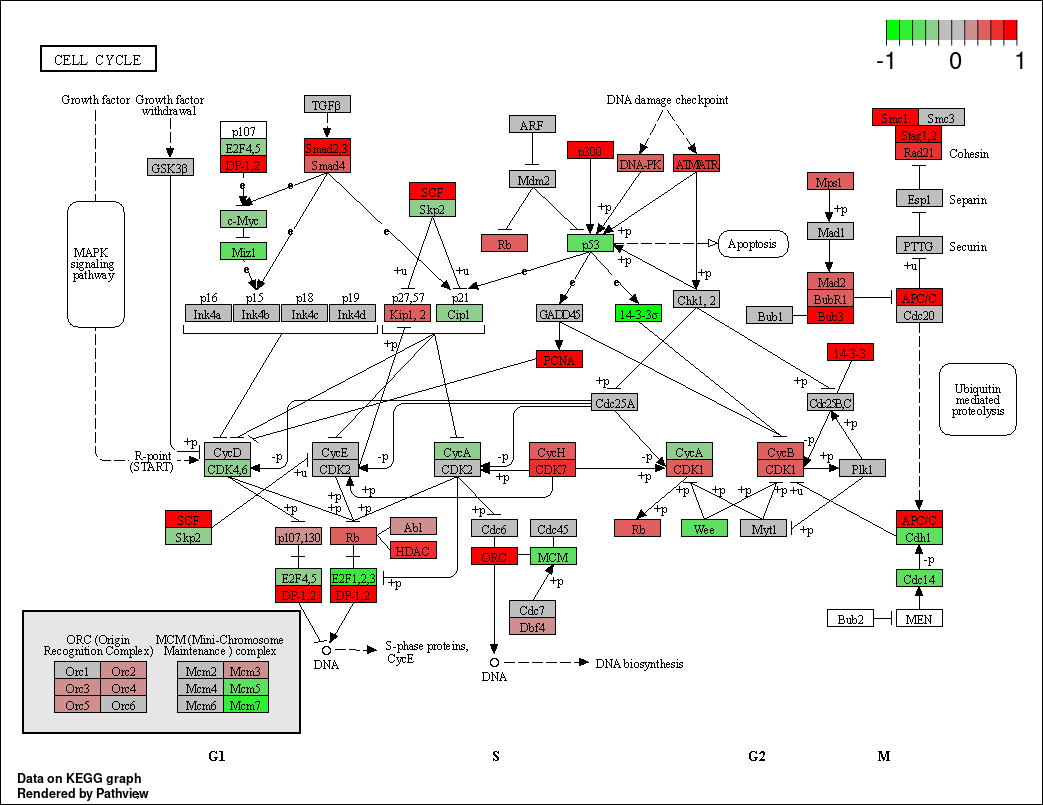
我们将主函数的结果赋值给了pv.out,我们不妨来看一下它的结构:
是一个长度为2的列表:
-
一个是plot.data.gene,储存画图用到的基因数据
一个是plot.data.cpd,储存的是药物或化合物节点的数据,我们之后会用到
plot.data.gene储存量我们画图的各种参数,是一个数据框。我们也可以对数据框中的数据做个性化的修改
看一下pv.out的组成结构
> str(pv.out)
List of 2
$ plot.data.gene:'data.frame': 92 obs. of 10 variables:
..$ kegg.names: chr [1:92] "1029" "51343" "4171" "4998" ...
..$ labels : chr [1:92] "CDKN2A" "FZR1" "MCM2" "ORC1" ...
..$ all.mapped: chr [1:92] "1029" "51343" "4171,4172,4173,4174,4175,4176" "4998,4999,5000,5001,23594,23595" ...
..$ type : chr [1:92] "gene" "gene" "gene" "gene" ...
..$ x : num [1:92] 532 919 553 494 919 919 188 432 123 77 ...
..$ y : num [1:92] 124 536 556 556 297 519 519 191 704 687 ...
..$ width : num [1:92] 46 46 46 46 46 46 46 46 46 46 ...
..$ height : num [1:92] 17 17 17 17 17 17 17 17 17 17 ...
..$ mol.data : num [1:92] 0.129 -0.404 -0.42 0.986 0.936 ...
..$ mol.col : chr [1:92] "#BEBEBE" "#5FDF5F" "#5FDF5F" "#FF0000" ...
$ plot.data.cpd : NULL4. 实例
除了上面普通的KEGG信号通路,Pathview包也可以在那些炫酷的KEGG通路图上进行映射,这里我们以人类的氧化磷酸化通路为例,我们先去到KEGG官网,查询得到氧化磷酸化通路的KEGG ID为00190,如下:
我们可以看到这幅通路图原本的样子就很好看
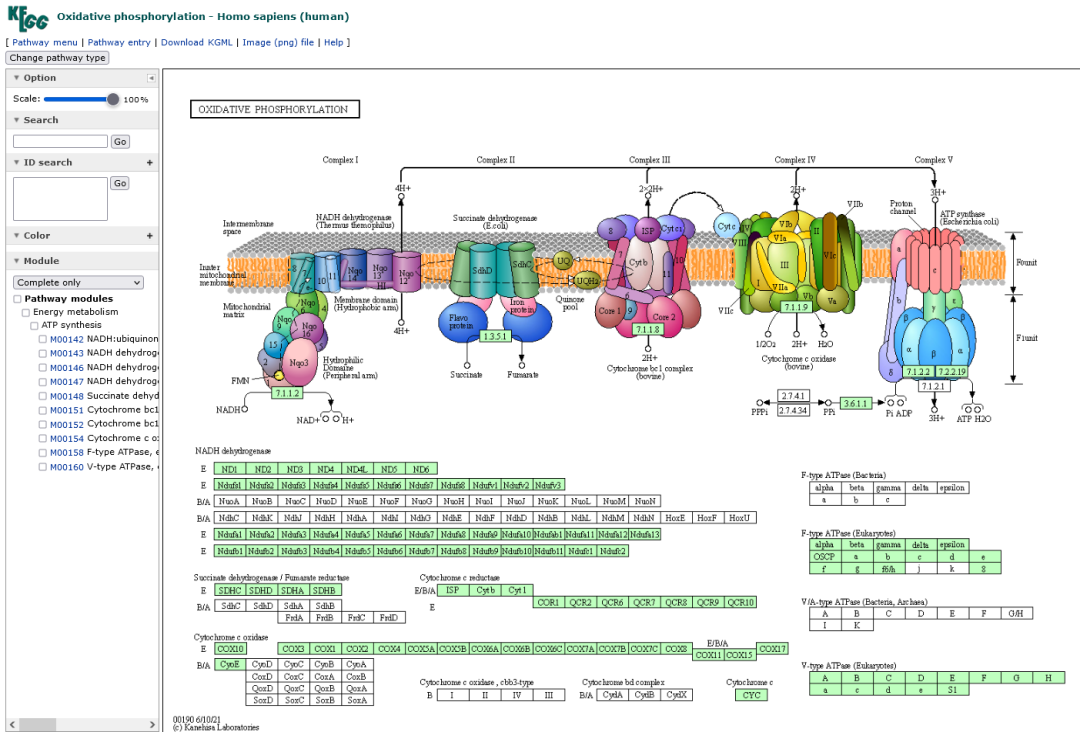
得到了通路ID后,我们就可以利用pathview()函数将我们的基因数据映射到通路图上啦。函数的参数不用变,只要改变pathway.id参数就可以了。
pv.out <- pathview(gene.data = gse16873.d[, 1],
pathway.id = "00190",
species = "hsa", #人类样本
out.suffix = "gse16873", #输出文件的后缀
kegg.native = TRUE)得到的结果如下,我们就将我们的单样本在该通路上的数据映射了上去: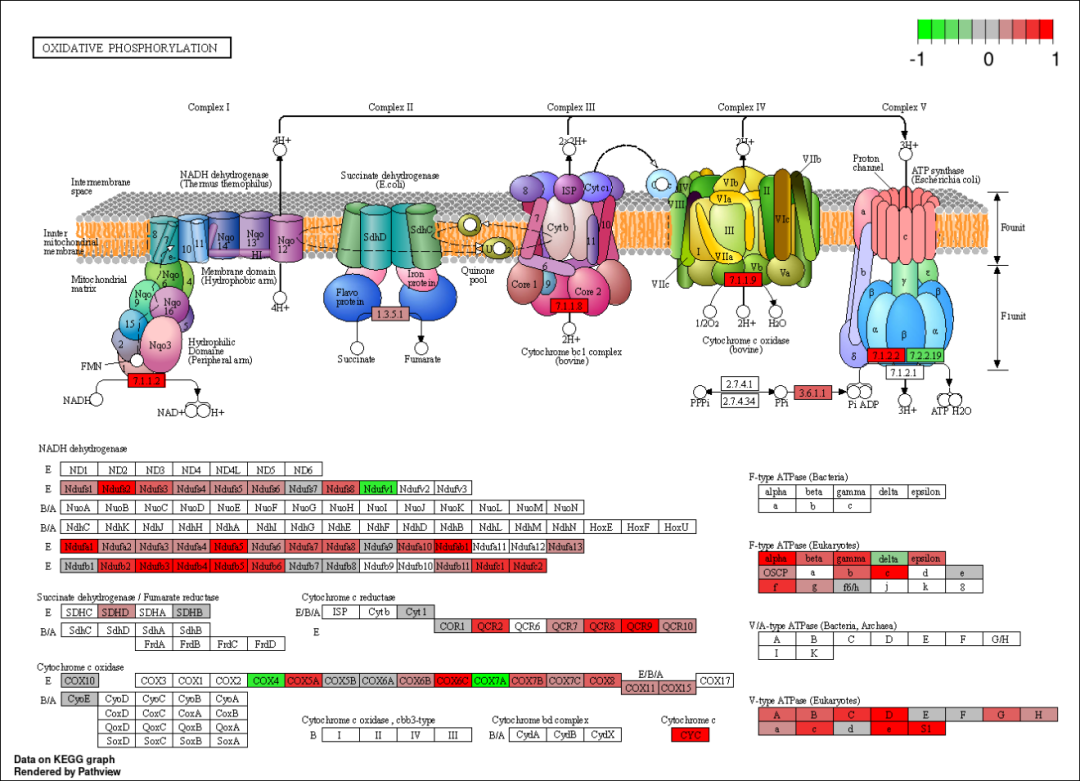
5. 小结
我们简单回顾一下,我们用到的主要函数便是pathview(),再提供了一个样本的基因数据和一个指定的KEGG通路的ID后,我们就得到了一副经典的KEGG通路图,并且通路图映射着该样本的基因表达量。