点击蓝字 关注我们
水产养殖模式对水产动物皮肤、口腔和肠道微生物群落组装及宿主适应性的影响

https://doi.org/10.1002/imt2.17

4.5
iMeta
RESEARCH ARTICLE
● 2022年4月5日,南昌大学丁霞等在iMeta在线发表题为“The impact of aquaculture system on the microbiome andgut metabolome of juvenile Chinese softshell turtle (Pelodiscus sinensis)”的研究性文章。
● 该研究聚焦水产动物微生物组,阐明在不同的水产养殖模式下宿主相关微生物的群落结构和构建机制,以及对水产动物的环境适应性影响,为更好的管理农业微生物服务可持续农业提供新的见解。
● 第一作者:丁霞(南昌大学)
● 通讯作者:丁霞
● 通讯作者:洪一江
摘 要
商业水生动物微生物组显著影响宿主在各种水产养殖系统中的成功养殖。然而,人们对此知之甚少。本研究比较了两种水产养殖系统,即稻渔养殖(RFC)和集约池塘养殖(IPC),以解构中华鳖幼鳖 (Pelodiscus sinensis)的皮肤、口腔和肠道微生物组以及肠道代谢组。我们结果表明,两种养殖系统下中华鳖的肝脏脂质代谢和体型有显著差异。与IPC相比,在RFC中的中华鳖微生物群落具有更高的α多样性和功能冗余。水产养殖系统对肠道微生物组的影响最大,其次是皮肤微生物组,最后是口腔微生物组。溯源追踪表明,与IPC相比,RFC中华鳖的皮肤、口腔和肠道微生物群落更多的来自于非自身身体部位之外的未知源。值得注意的是,RFC中华鳖口腔和皮肤微生物组具有更高比例的泛化种和更广泛的生态位广度,但肠道微生物组没有这种范式。零模型分析表明,与IPC相比,中华鳖的口腔和皮肤微生物群落组装在RFC养殖系统中受确定性过程的比例显著增加,但肠道没有这种范式。我们进一步分析得到对五种差异肠道代谢物(2-酮戊二酸、N-乙酰-D-甘露糖胺、顺式-4-羟基-D-脯氨酸、烟酰胺和L-丙氨酸)贡献最大的关键基因和微生物,这些差异肠道代谢物属于氨基酸代谢、GABA能突触、ABC转运蛋白、不饱和脂肪酸的生物合成以及柠檬酸循环等代谢路径。我们的研究结果为水产养殖系统如何影响水生动物微生物群落结构和组装机制提供了新的见解,也突出了关键微生物和基因对肠道微生物组-代谢组关联中代谢物差异的贡献。
关键词:水产养殖系统,水生动物,宿主相关微生物组,宿主-微生物组相互作用,微生物组-代谢组关联
亮 点

合理的农业养殖模式对全球生物多样性、生态系统功能和可持续农业至关重要。农业养殖模式如何影响农田微生物群落结构和功能,从而影响养殖动物的适应性研究不多。本研究聚焦水产动物微生物组,阐明在不同的水产养殖模式下宿主相关微生物的群落结构和构建机制,以及对水产动物的环境适应性影响,为更好的管理农业微生物服务可持续农业提供新的见解。
● 水产养殖环境显著影响中华鳖的表型、微生物群落组装和肠道代谢物
● 不同的水产养殖系统塑造了组织特异性的微生物群落组装,并部分通过肠道代谢物改变宿主的适应性
● 外源微生物输入和群落组装过程驱动了养殖系统中微生物多样性的产生和维持
● 识别了五个差异肠道代谢物的关键基因和微生物贡献者
视频解读
Bilibili:https://www.bilibili.com/video/BV1oB4y1m7Y1/
Youtube:https://youtu.be/91oOEcaW14Y
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
鱼类和其他水产食品(蓝色食品)为全球32亿人口提供蛋白质及营养,但它们对粮食安全的贡献却被忽视了。其中,仅小规模渔民和水产养殖 (SSFA) 就为全球约 10 亿人提供食物。目前,水产养殖面临着气候变化、环境影响和人口增长等前所未有的挑战。农业实践可能会重塑流域结构,影响水生动物的健康。例如,高密度集约化池塘养殖(IPC)极大地减少了人类赖以生存的生物多样性,并导致环境问题和病害发生。因此,迫切需要发展低投入、低影响的水产养殖模式。农业-水产养殖一体化(IAA)是一种在同一地点同时生产谷物和动物蛋白的农业养殖方法,高度重视生态环境并促进可持续性。稻渔养殖 (RFC) 是世界上最受欢迎的 IAA 系统之一,特别是在热带和亚热带亚洲。RFC在稻田中养殖水生动物,同时种植水稻(图 1A)。其实RFC 实践在亚洲已有 1700 多年的传统和历史。就RFC的总体规模而言,中国是主要生产国,RFC现在是中国最重要的农业系统之一。稻渔养殖系统养殖的水产动物包括鱼、中华鳖、鲤鱼、虾和青蛙等。
水生动物体内蕴藏着数以万亿的微生物,这些微生物随环境改变具有显著动态变化的组成和功能。大量证据揭示了动物与其相关微生物群之间相互作用的重要性。不同的身体部位,如肠道、口腔和皮肤,具有不同的微生物群落。宿主相关的微生物组为宿主的生理和适应性提供了基本功能。研究表明肠道微生物群有助于宿主营养吸收、免疫系统发展和肠道发育。与肠道中的微生物组类似,皮肤微生物在保护宿主免受病原体入侵和训练宿主免疫系统方面发挥重要作用。口腔微生物群在牙周炎、龋齿和全身性疾病中同样发挥至关重要的作用。一方面,微生物对水生动物的健康养殖至关重要,可以通过人工添加以发挥其作用。另一方面,在高密度饲养模式中有效控制水产养殖病原微生物群至关重要。因此,了解和操纵水产养殖环境中的微生物群落具有巨大的商业潜力。然而,我们目前对水产养殖微生物群和这些系统的整体生态学缺乏了解,阻碍了水产养殖环境中微生物的成功管理。
中华鳖 (Pelodiscus sinensis)是亚洲最具商业价值的水生物种之一。在亚洲,尤其是中国、日本和韩国,中华鳖具有很高的营养和药用价值。与 IPC 系统相比,人们普遍认为 RFC 有利于维护生物多样性,并生产高营养价值的水产品。因此,有必要系统地评价水产养殖系统对中华鳖和环境的影响。此外,在系统发育上中华鳖位于amniotic动物(鱼类和两栖动物)和amniotic动物(鸟类和哺乳动物)之间,在脊椎动物进化中起着至关重要的作用。因此中华鳖是一种遗传进化、系统发育和生物多样性的良好模型动物。然而,很少对来自同一水生动物个体的肠道、口腔和皮肤微生物组进行系统的比较分析,而且这些与宿主相关的组织特异性的微生物组是否会在相同的空间尺度上响应相似的环境而发生相似的变化,这在很大程度上是未知的。
实验方案
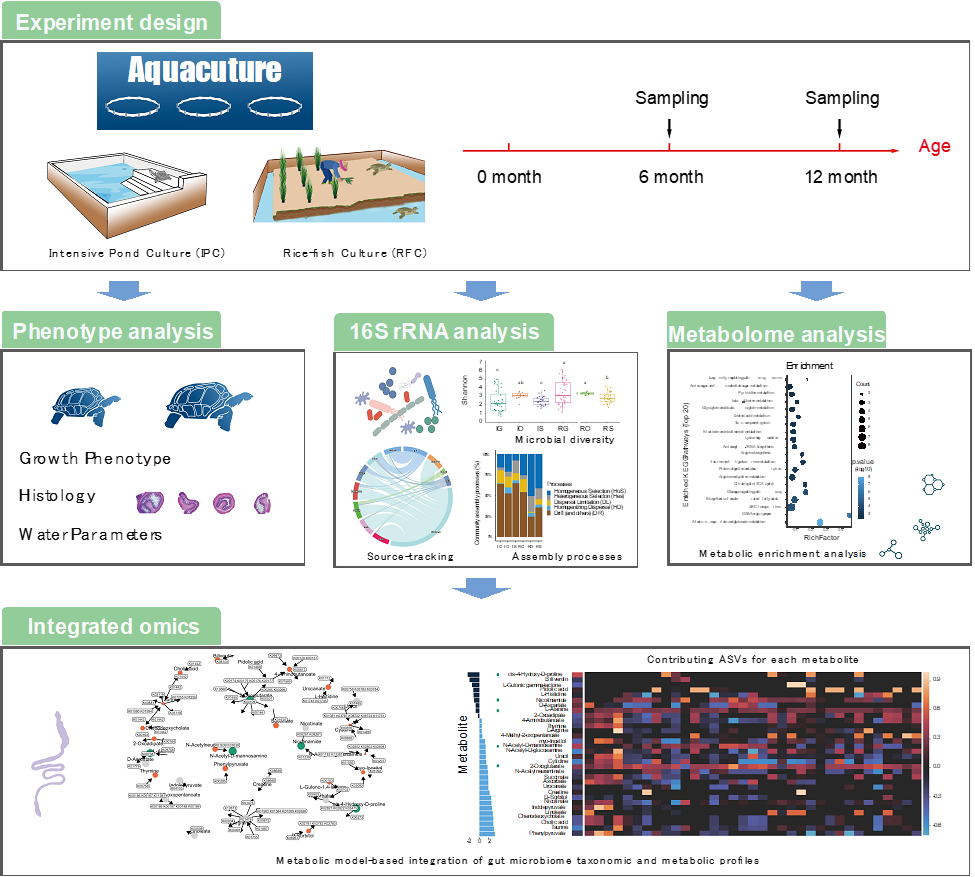
中华鳖幼苗在温室(GC)养殖到6个月大,将其从GC分别转移至集约化池塘(IPC)和稻渔生态养殖田(RFC)(图1A, 表S1)。我们分别取样温室养殖的6个月大中华鳖,以及池塘与稻渔生态田养殖的12个月大中华鳖。对其皮肤(腹部,脚,背部),口腔和肠道(十二指肠,回肠,结肠,直肠和整个肠道)进行取样。检测水体理化性质,中华鳖的表型(生长表型,肠道和肝脏免疫组化),16s rRNA基因扩增子测序分析皮肤、口腔和肠道的微生物多样性,LC-MS检测肠道代谢组。
结 果
中华鳖微生物群落结构
通过肠道免疫组化分析将中华鳖肠道分为四节:十二指肠,回肠,结肠,直肠(图S1)。240 个样本中总共获得了 23,184 个特征和 10,983,297 个reads,平均read数为 45,763(范围:22,000-55,039)。不同身体部位的微生物群落组成差异显著(Kruskal-Wallis 检验,p < 0.05;图 1D)。在口腔、腹部、四肢和背部分别检测到 2500、2481、2681、2192 个 ASVs,这四个部位共享 837 个ASVs。在十二指肠、回肠、结肠和直肠内分别检测到 9102、5708、4851 和 2561 个 ASVs,这四个部位之间共享了 761 个 ASVs。共享的 ASVs 分别占皮肤/口腔和肠道微生物相对丰度的 14.4% 和 4.8% (图 S2)。
在门水平,皮肤(背部、四肢和腹部)和口腔以变形菌门、拟杆菌门、放线菌门和热菌门为主,肠道(十二指肠、回肠、结肠和直肠)以变形菌门、拟杆菌门、厚壁菌门和梭杆菌为主(图 1D 和表 S2)。在属水平,皮肤(背部、四肢和腹部)和口腔以不动杆菌属、气单胞菌属和罗氏菌属为主,肠道(十二指肠、回肠、结肠和直肠)以梭菌属、螺杆菌属、鲸蜡杆菌属、拟杆菌属和气单胞菌为主(图 1D 和表 S3)。微生物各个属在不同身体区域、年龄和水产养殖系统中存在显著差异(Kruskal-Wallis 检验,p < 0.05;图 1E 和表 S4)。然而,皮肤(背部、四肢和腹部)和肠道(十二指肠、回肠、结肠和直肠)内不同部位的微生物组间没有显著差异(Kruskal-Wallis 检验,p > 0.05)。在口腔和皮肤中,RFC 和 IPC 的微生物组中不动杆菌的比例显著高于 GC(Kruskal-Wallis 检验,p < 0.05)。GC 和 IPC 的微生物组中气单胞菌的比例明显高于 RFC(Kruskal-Wallis,p < 0.01)。在肠道中,IPC 的微生物组中螺杆菌的比例最大,GC 的微生物组中梭菌的比例最大(Kruskal-Wallis 检验,p < 0.01)。
Bray-Curtis 距离的主坐标分析 (PCoA)结果表明,年龄、养殖环境和身体部位均将微生物组显著区分开(Adonis,p < 0.001,图 1F)。置换多变量方差分析(PERMANOVA)表明,身体区域是微生物组数据中最大的变异源(24.6%,p < 0.001),养殖培养模型是第二个变异源(16.2%,p < 0.001),年龄是第三个变异源(9.8%,p < 0.001;表 S5)。因此接下来,我们将分析养殖模型如何影响组织特异性的微生物群落。
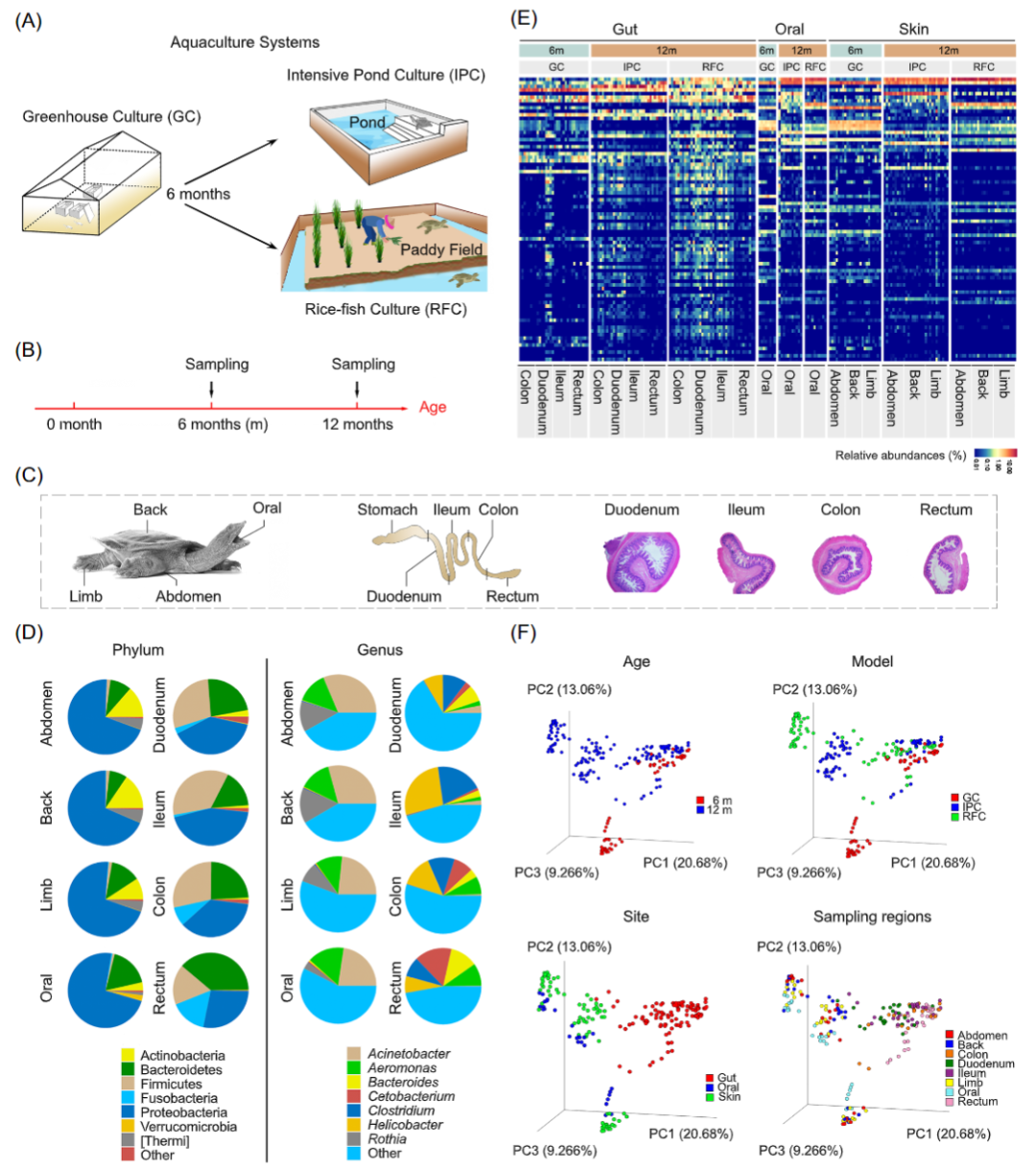
图1 中华鳖皮肤、口腔和肠道的微生物群落组成
(A) 两种不同水产养殖系统的示意图。(B) 采样时间方案。(C) 皮肤、口腔和肠道等采样部位。(D) 在门和属水平,每个身体部位微生物相对丰度的饼图。(E) 前 80 个属物种在不同养殖模式、年龄、身体部位的相对丰度分布。(F) 基于 Bray-Curtis 距离的主坐标分析(PCoA)。样本按年龄、养殖模型、身体部位和采样区域着色。GC,温室培养系统;IPC,集约化池塘养殖;RFC,稻鱼养殖。
RFC 和 IPC 中微生物组α 和 β 多样性的比较
为了比较组织特异性微生物组对两种水产养殖环境的响应,分析了中华鳖皮肤、口腔和肠道微生物组 α 和 β 多样性的变化。RFC 中微生物群落 α多样性在肠道和皮肤区域显著高于 IPC中微生物群落(Kruskal-Wallis 检验,p < 0.05),但口腔区域没有显著差异(Kruskal- Wallis 检验,p > 0.05;图 2A)。在 RFC 和 IPC 两个养殖系统中,肠道微生物组的 α 多样性均显著高于口腔和皮肤微生物组(Kruskal-Wallis 检验,p < 0.05;图 2A)。
微生物群落结构的β 多样性分析表明, IPC 与RFC养殖模式能将微生物组样本在第二个 PCoA 轴上明显分离(PERMANOVA,p < 0.05;图 2B)。不同部位微生物组均与其他组具有显著差异(成对 PERMANOVA检测,p < 0.05;表 S6 和图 2B)。用Bray-Curtis 和 Jaccard 计算相异指数,RFC 的肠道微生物群落比 IPC 的组内具有更大的相异指数。并且,在两种养殖模型中,肠道微生物群落的组内相异指数均大于口腔和皮肤微生物群落(图 2C)。
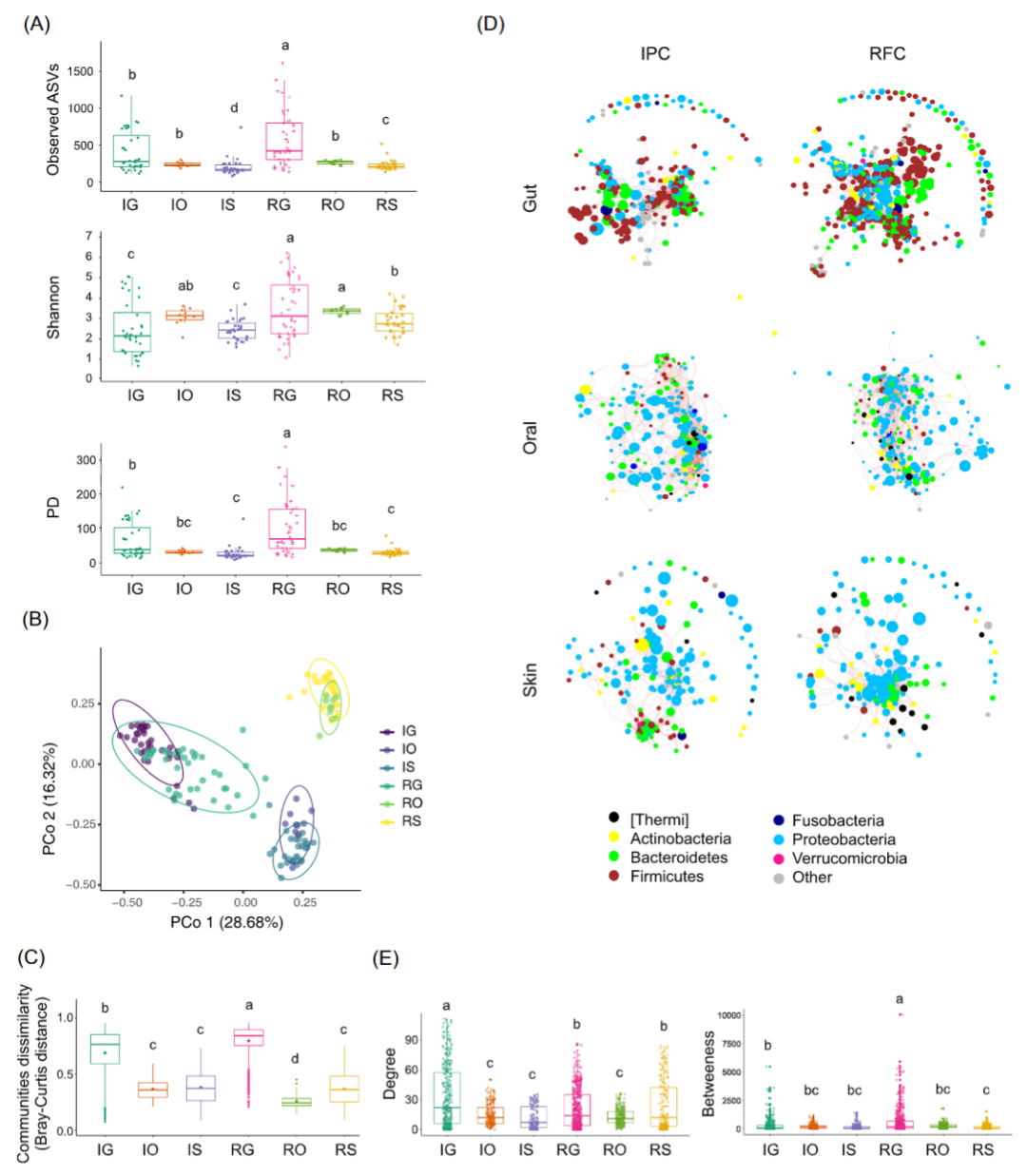
图2 RFC 和IPC 中的微生物多样性和群落共现网络
(A) α 多样性指数。(B)β 多样性分析。PCoA 基于Bray-Curtis 相异距离。(C) 微生物群落相异指数。(D) 微生物群落共现网络 (p < 0.5 和abs (r) > 0.6)。网络节点根据 ASV 的相对丰度(log10 转换的相对丰度),按门和大小着色。边缘的负相关用蓝色表示,边缘的正相关用红色表示。(E) 微生物群落共现网络的节点边统计。显著差异(Kruskal-Wallis检验,p< 0.05)用不同的字母表示。IG,IPC中的肠道;IO,IPC中的口腔区域;IS,IPC中的皮肤;RG,RFC 中的肠道;RO,RFC 中的一个口头区域;RS,RFC 中的皮肤。
RFC 和 IPC 中微生物组的共线性网络分析
与皮肤和口腔微生物网络相比,肠道共线性网络具有更多的边和顶点数(表 S7)。对于肠道和皮肤区域,RFC 和 IPC 之间的网络度存在显著不同,但口腔微生物没有显著差异(Kruskal-Wallis 检验,p < 0.05;图 2D,E)。RFC 和 IPC 之间的网络介数在肠道中存在显著差异,但在口腔和皮肤中没有显著差异(Kruskal-Wallis 检验,p < 0.05;图 2D,E)。对于肠道和皮肤区域,RFC 的负相关数显著高于 IPC(Wilcoxon 检验,p < 0.05;图 2D 和表 S7)。此外,还观察了网络中心点如何响应养殖模型。对于皮肤区域,IPC 中的网络中心以拟杆菌门和厚壁菌门为主,而 RFC 中的网络中心以变形菌门为主(图 S3)。
总之,微生物群落多样性和共线性网络分析结果表明,水产养殖系统对肠道微生物组群落结构的影响最大,其次是皮肤微生物组,最后是口腔微生物组。
RFC 和 IPC 之间肠道、口腔和皮肤微生物组的分类和功能差异
LEfSe分析表明,在肠道,厚壁菌门、拟杆菌门、放线菌门、Beta变形菌门、乳杆菌目伯克霍尔德菌目和假单胞菌目、毛螺菌科和莫拉菌科、乳杆菌属、梭菌属、不动杆菌属、肠球菌属、罗氏菌属、鞘氨醇单胞菌属、瘤胃球菌属的相对丰度、罗氏菌属、普氏杆菌、双歧杆菌、真杆菌、粪杆菌、类杆菌和 气单胞菌在 RFC中大量富集(LEfSe,p<0.05),变形杆菌、梭杆菌、螺杆菌、醋酸杆菌、甲基杆菌在IPC中大量富集(LEfSe,p < 0.05;图 3A, S4)。在口腔,变形杆菌门、伯克氏菌目和假单胞菌目、莫拉菌科、不动杆菌属在RFC中富集(LEfSe,p<0.05),气单胞菌目和肠杆菌目、气单胞菌属在IPC中大量富集(LEfSe,p<0.05;图3B)。在皮肤,伯克氏菌目和假单胞菌目、莫拉菌科、不动杆菌属在RFC中高度富集(LEfSe,p<0.05),微球菌科和罗思氏菌属和气单胞菌属在IPC中富集(LEfSe,p<0.05;图3C)。据观察,伯克氏菌目和假单胞菌目、莫拉菌科、不动杆菌属是 RFC 中肠道、口腔和皮肤区域的共有富集物种。
为了了解微生物群落功能冗余的差异,用Tax4Fun2计算功能冗余指数(FRI)。在肠道、口腔和皮肤中,RFC的FRI均明显高于IPC (图3DF)。特别是, RFC和IPC之间的FRI在肠道微生物群落中具有最显著的差异。结果表明,在所有肠道、口腔和皮肤微生物群落,尤其是肠道微生物群落中,RFC系统中的微生物群落比 IPC更加功能冗余。
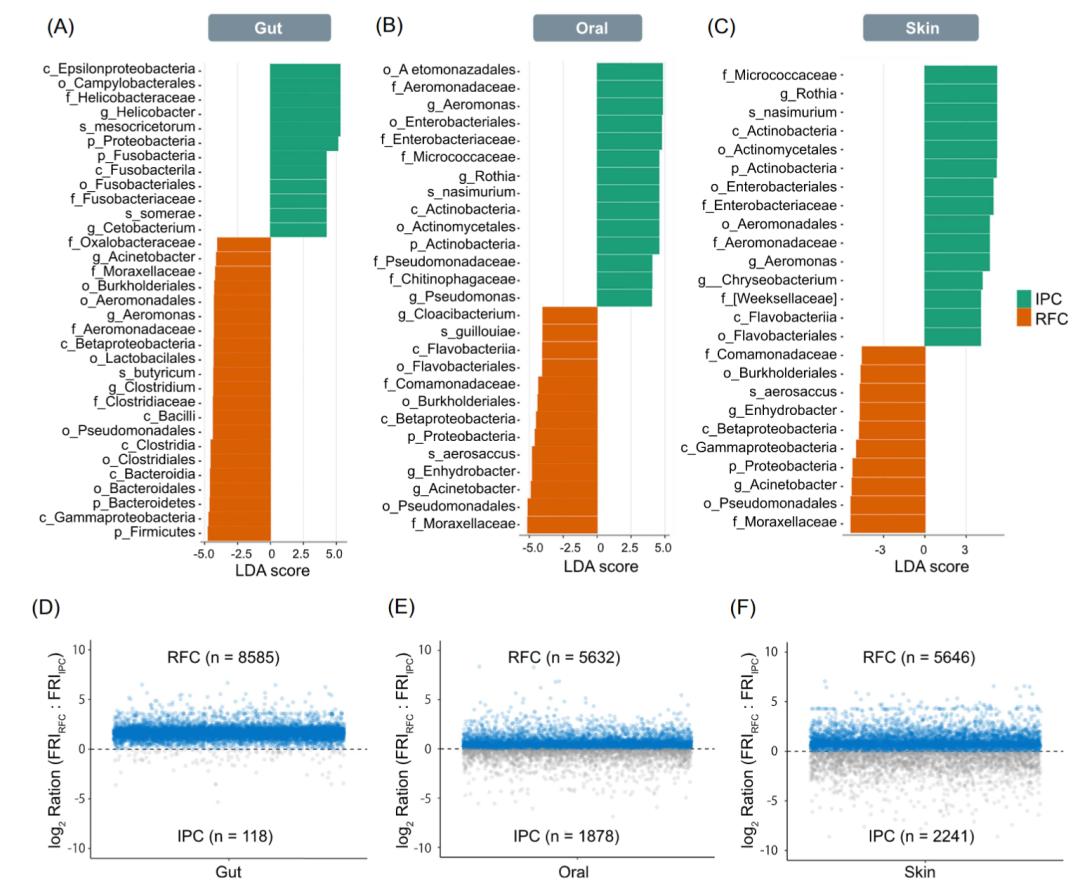
图3 RFC 和IPC 之间皮肤、口腔和肠道微生物组的分类和功能差异
(A-C) LEfSe分析RFC和IPC 之间 (A) 皮肤、(B)口腔和(C) 肠道的显著差异微生物。LDA 分数>4 和 p < 0.05的被展示。(D-F)比较RFC 和 IPC 之间肠道(D)、口腔 (E) 和皮肤(F) 微生物群落的功能基因冗余。高 FRI 表明特定功能在所有群落中几乎无处不在。相反,低FRI 表明该功能存在于少数密切相关的物种中。log比值大于0 表示在 RFC 中冗余度较高,log比值小于0 表示在 IPC 中冗余度较高。所有预测均使用97% 的相似性阈值进行。
溯源分析
前面分析表明RFC中的微生物群落与IPC相比表现出显著更高的α多样性和功能冗余,为了分析多样性差异的来源,我们使用了动态贝叶斯推理神经信息流网络进行溯源分析。将6 个月大的中华鳖皮肤、口腔和肠道定义为来源。结果表明,12个月大的中华鳖微生物群落来源于多种环境,包括6 个月大的中华鳖的皮肤、口腔、肠道和未知源,并且未知源是RFC和IPC微生物群落的主要来源(图4A和表S8)。未知源可能是周围的水体、土壤和环境。在RFC和IPC中,肠道微生物组均主要来源于肠道和未知源,部分来源于皮肤和口腔。在RFC和IPC中,皮肤和口腔微生物组均主要来源于皮肤、口腔和未知源,但IPC中的一小部分皮肤和口腔微生物组也来源于肠道(图 4B 和表 S8)。未知源的比例最高的是皮肤微生物组,其次是口腔微生物组,最后是肠道微生物组。特别注意的是,RFC 中的微生物群落比 IPC 更多的来源于未知源(图 4B 和表 S8)。
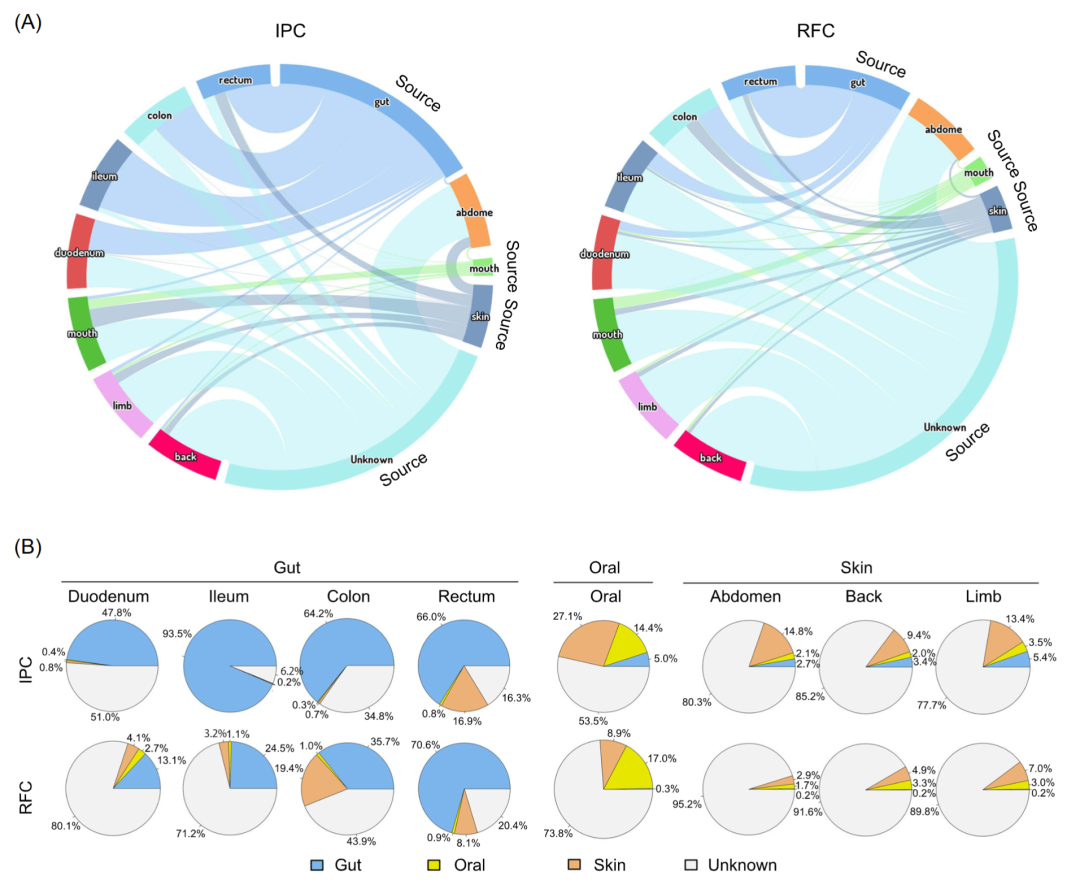
图4 溯源分析
(A) 目的样本与来源样本之间的微生物流。(B) 目的样本中不同来源占比估计。
微生物群落组装机制
将微生物群落被划分为泛化种和特化种进行分析。结果发现,在 RFC 和 IPC 中,口腔和皮肤具有更高比例的泛化种(图 5A)。在RFC 和 IPC 之间,肠道的泛化种(10.49% 对 8.63%)和特化种(74.17% 对 75.97%)的比例相当。然而,RFC 的口腔和皮肤泛化种占总多样性的 32.83% 和 24.77%,远高于 IPC,后者仅占 15.29% 和 15.52%。与之一致地,口腔和皮肤微生物群落的平均群落生态位宽度 (Bcom) 在 RFC 和 IPC 中明显大于肠道微生物群落(Kruskal-Wallis 检验,p < 0.05;图 5B)。并且RFC 的口腔和皮肤微生物群落的平均 Bcom 显著高于 IPC(Kruskal-Wallis 检验,p < 0.05),但 RFC 的肠道微生物群落的平均 Bcom 略窄于 IPC( Kruskal-Wallis 检验,p < 0.05)。结果表明,口腔和皮肤微生物组在群落水平上比肠道微生物组在代谢上更灵活。此外,与 IPC 相比,RFC 的口腔和皮肤微生物组在群落水平的代谢更灵活。
我们量化了确定性过程(均质和异质选择)和随机过程(分散限制、均质分散和漂移)如何影响皮肤、口腔和肠道微生物群落组装。根据不同部位对群落组装过程差异分为两种模式。对于皮肤和口腔,漂移是主导 IPC 中口腔(52.3%)和皮肤(64.6%)微生物群落的最重要过程。相比之下,均质选择是 RFC 中主导口腔 (51.0%) 和皮肤 (41.6%) 微生物群落的最重要过程。对于肠道,漂移是 IPC 和 RFC 中主导肠道微生物群落的最关键过程,平均相对重要性为 63.5% 和 53.6%。此外,观察到 RFC 的口腔和皮肤微生物群落比 IPC受到更大比例的确定性过程控制,而RFC 的肠道微生物群落比IPC受确定性过程的比例较小。
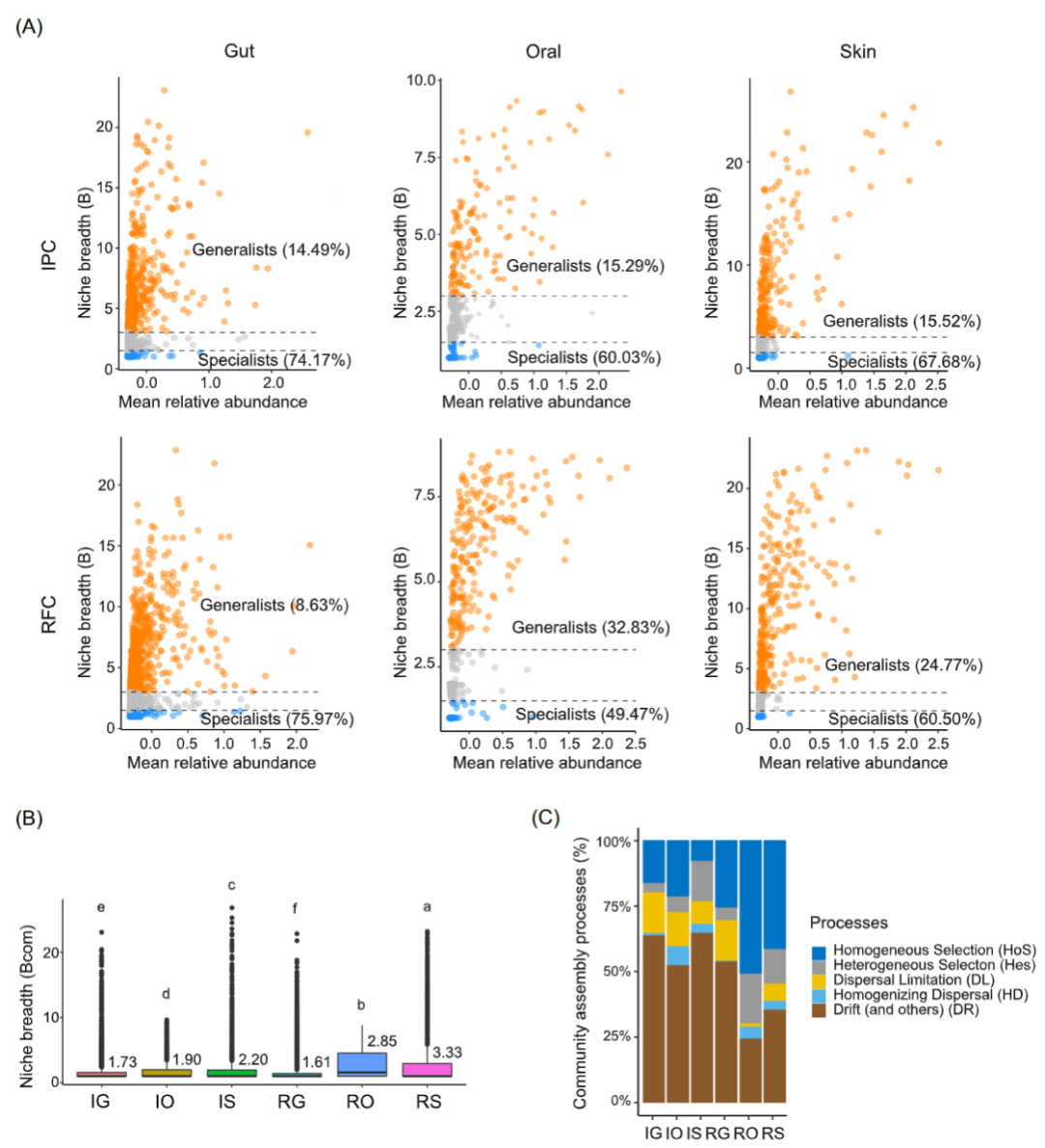
图5 微生物群落组装过程
(A)微生物组ASVs 的平均丰度与生态位宽度的关系。泛化种ASVs 为橙色,特化种 ASVs 为蓝色。(B)平均生态位宽度(Bcom) 的箱线图。显著差异(Kruskal-Wallis 检验,p< 0.05)用不同的字母表示。(C) RFC 和IPC 中皮肤、口腔和肠道微生物群落组装受确定性过程(均质和异质选择)和随机过程(分散限制、均质分散和漂移)主导百分比。
肠道微生物组-代谢组整合分析差异代谢物的关键微生物和基因贡献
代谢物集富集分析(MSEA) 和代谢途径分析(MetPA)鉴定了 82 个显著差异的肠道代谢物 (VIP > 1,t检测,p < 0.05;图 S5)。RFC 的肠道中存在较高浓度的脂肪酸和轭合物、嘧啶、嘌呤、TCA 酸、吡啶和单糖(Fisher 精确检验,p < 0.001;图 6A 和表 S10)。IPC 肠道中的氨基酸和肽浓度较高(Fisher 精确检验,p < 0.001;图 6B 和表 S10)。差异代谢通路涉及丙氨酸、天冬氨酸和谷氨酸代谢、GABA能突触、不饱和脂肪酸的生物合成、胰高血糖素信号通路、柠檬酸循环(TCA循环)、精氨酸和脯氨酸代谢(图6C和表S11)。
使用基于模型的代谢组和物种丰度整合分析(MIMOSA)研究肠道内容物中微生物组和代谢组之间的关系。总体而言,32个差异代谢物中,5个与代谢潜能显著正相关(15.6%),16个显著负相关(50%)。预测良好(正相关)的代谢物包括2-酮戊二酸、N-乙酰基-D-甘露糖胺、顺式-4-羟基-D-脯氨酸、烟酰胺和L-丙氨酸。计算群落代谢潜能(CMP) 分数以识别每种代谢物的潜在基因、反应和物种贡献者,大多数代谢物有几个关键基因贡献者,这些基因贡献者编码催化代谢物产生和/或降解的酶(图6D 和表 S12)。具体来说,这些预测良好的代谢物与氨基酸代谢、辅因子和维生素代谢以及能量代谢相关,这些是肠道微生物催化过程的基本类别(图S6)。关键物种贡献者分析表明,Beta变形菌门、消化链球菌科、瘤胃球菌科、气单胞菌科和毛螺菌科以及拟杆菌属和假单胞菌属的ASV 促进了 2-氧戊二酸的合成,消化链球菌科和红螺菌科的ASV 促进了 N -乙酰基-D-甘露糖胺(图6E 和表 S13,14)。此外,粘球菌、链霉菌科和红环菌科、诺卡氏菌属的ASV 促进了顺式-4-羟基-D-脯氨酸的降解,消化链球菌科和毛螺菌科的ASV 促进了烟酰胺的降解。
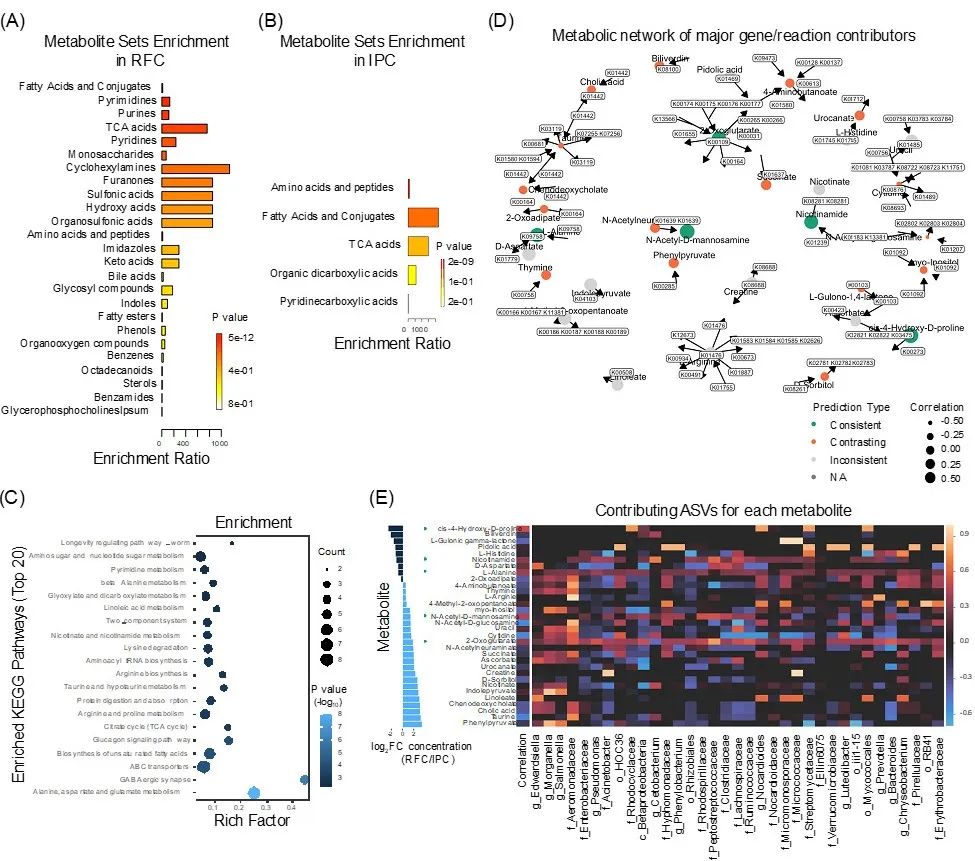
图6 基于代谢模型的肠道微生物组和代谢组整合分析
(A,B) 代谢物富集分析(MSEA)。(C) 基于 KEGG 完整库的代谢富集分析。(D)主要基因/反应的代谢网络展示每种代谢物的关键贡献者。每个节点的颜色和大小表明该代谢物被群落代谢潜力预测的程度。良好预测的代谢物定义为CMP 分数的变化与 FDR < 0.01 时测量的代谢物丰度变化显著相关(使用Mantel 检验)的代谢物。反预测代谢物同样定义为 CMP 评分变化与测量的代谢物丰度变化显著负相关(FDR< 0.01)的代谢物。(E) MIMOSA 模型预测参与特定代谢物生产或降解的微生物。所有代谢物的潜在分类贡献者的热图可视化。用绿色圆圈标记的代谢物表示预测的一致类型。CMP,群落代谢潜力。
中华鳖的表型性状和养殖环境理化性质
为了比较养殖环境对微生物群-宿主-环境相互作用的影响,检测了中华鳖的生长性状和组织学特征,以及RFC和IPC中水的物理参数。两种水产养殖系统之间的中华鳖生长相关表型性状存在显著差异(t检验,p< 0.05),RFC 中中华鳖的发育明显慢于IPC(图 7A)。RFC显著促进了肝脏脂肪的积累(t检验,p< 0.001;图 7B,C)。而肠道(十二指肠、回肠、结肠和直肠)组织没有显著的组织学变化(t检验,p> 0.05),除了RFC中十二指肠组织中肠绒毛的长度明显大于IPC(t检验,p< 0.05;图 7D-G)。此外,观察到RFC 和 IPC 之间的环境因素(例如,pH、DO、TOC、TON和NO3-)和水的浮游生物没有显著差异(t 检验,p> 0.05;图 S7 和表S15)。
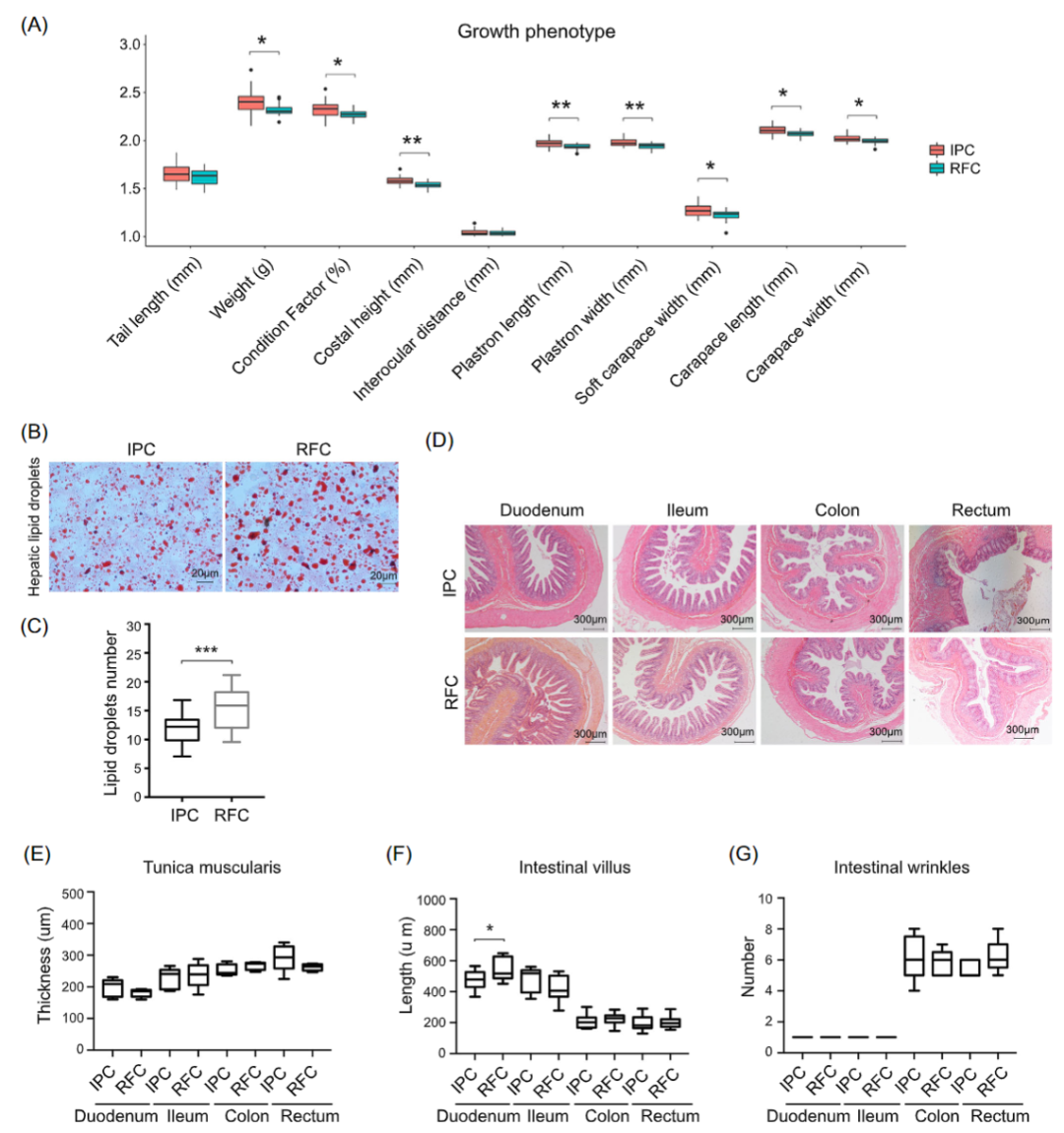
图7 RFC 和 IPC 对中华鳖的生长条件和表型性状影响
(A)两种农业培养体系中中华鳖的生长相关表型性状。数据经过log10 转换。(B) 肝脂滴的代表性油红O 染色。比例尺:20 μm。(C)肝脂滴的数量统计。(D)肠道(十二指肠、回肠、结肠和直肠)的代表性H&E 染色。比例尺:300 μm。(E)肌层厚度。(F)肠绒毛的长度。(G)肠道皱纹数。*p< 0.05、**p < 0.01 和***p < 0.001(t 检验)。
讨 论
两种养殖模式下微生物的多样性和稳定性
有报道观察到稻田的肠道微生物组比池塘的肠道微生物组具有相对更高的丰富度。我们的实验结果也得到类似结论,RFC 中微生物群落的 α多样性明显高于 IPC(图 2A)。我们探讨了RFC中微生物群落的 α多样性更高的产生和维持机制。在 IPC 中,相对均匀的水文条件导致相对较低的 α多样性。RFC 中复杂的空间结构(比如稻田、水渠、水稻)导致了更高的 α多样性。这种推测也得到了溯源跟踪分析结果的支持。我们发现在RFC 中的微生物群落比 IPC 起源于更多未知源(图 4),这表明 RFC 中的中华鳖从更复杂的稻田周围环境中获得了更多的微生物来源。因此,稻田养殖模式下(RFC)中华鳖与各种周围环境的更丰富的相互塑造出了微生物群落更高α多样。
微生物生态系统的稳定性被认为对宿主的健康至关重要,但人们对此知之甚少。人们普遍认为,生物多样性与稳定性之间存在正相关关系。我们的数据显示,RFC 中中华鳖微生物群落比 IPC 显示出更高的 α多样性(图 2A),推测 RFC 的微生物组比 IPC 的微生物组更稳定。然而,一项使用数学模型研究多样性和稳定性之间关系的研究显示出相互矛盾的结果,尤其是健康和病理状态之间的多样性比较。因此,微生物多样性的测量只是一个起点,而不是群落结构结构的“答案”。因此我们扩展了方法,使用网络交互和功能冗余来深入分析我们的微生物组数据。Coyte 等人的模型预测,认为引入网络负反馈回路的生态竞争将增强微生物群落的稳定性。在中华鳖肠道和皮肤区域,RFC 的网络负相关数显著高于 IPC(图 2D 和表 S7)。此外,最近的工作表明功能冗余对诱导肠道微生物群稳定性的重要性。RFC 中的中华鳖微生物群落比 IPC 显示出更高的功能冗余,尤其是肠道微生物群落(图 3D-F)。总之,我们的证据证明 RFC 可能通过促进宿主-微生物组的稳定性来有益于宿主的适应性。
两种养殖模式中微生物群落组装机制
微生物群落组装对于了解微生物群落调节生态系统功能的机制至关重要。RFC 比 IPC的微生物群落从周围环境(如水生和土壤环境、水稻和食物)中获得更多异质物种,导致更高的 α 多样性和功能冗余(图 2A 和 3D-F)。中性群落模型的结果进一步支持这一观点,在 RFC 中观察到更高确定性的组装过程,特别是在皮肤和口腔中(图 5C)。在具有更异质的环境系统中,确定性过程可能会主导作用。因为 IPC中的池塘水流和相对均质的水文条件会导致更高水平的微生物扩散,随机性过程占主导。而RFC中复杂的空间结构和相对异质的空间条件会导致更高水平的微生物环境选择,确定性过程占主导。
根据生存策略,微生物被分为泛化种和特化种。与肠道微生物群落相比,口腔和皮肤微生物群落的泛化种比例和生态位宽度明显更高(图 5A)。因此,推测皮肤和口腔微生物组在新陈代谢方面更具灵活性,能够适应更广泛的环境,也比肠道微生物组更能抵抗环境影响。众所周知,皮肤和口腔区域是开放区域,但肠道区域是厌氧封闭区域,其中微生物组受到电子受体可用性的强烈驱动。肠道区域的低溶解氧导致特化种对环境的强烈过滤,所以肠道中观察到的特化种比例显著高于口腔和皮肤(图 5A)。人们认为,泛化种比特化种更代谢多样,但效率较低,而特化种比泛化种更有效,但代谢活性更低。肠道是身体最活跃的代谢区域,需要高效的微生物组来合成和降解代谢物。因此,肠道拥有更高比例的特化种来执行受限但高效的新陈代谢,而皮肤和口腔拥有更高比例的具有广泛代谢能力以适应广泛环境条件的泛化种。
与 IPC 相比,RFC 中的口腔和皮肤微生物群落拥有更高比例的泛化种和更少的特化种(图 5A)。与之一致的,RFC 中的皮肤和口腔微生物群落表现出比 IPC 更宽的生态位广度(图 5B)。人们普遍认为,具有更广泛生态位宽度的泛化种在群落水平上更具新陈代谢灵活性并适应更广泛的环境。一个可能的解释是,RFC 是一个比 IPC 更异质的环境,导致更多的未知环境来源和更多的环境过滤,最终导致更高的 α 多样性和代谢灵活的微生物群落以适应多样化的异质环境。此外,泛化种和特化种的微生物组装由不同的过程控制。许多研究发现,泛化种和特化种的组装分别受到中性过程和环境选择的调控。但我们的数据表明,与 IPC 相比,RFC 的口腔和皮肤微生物组的泛化种比例更高。尽管如此,同质选择仍然是 RFC 中调控口腔和皮肤微生物群落的最关键过程(图 5)。先前的结论是基于自由生活的微生物组。这一有争议的发现仍然是一个悬而未决的问题,即组装过程如何影响宿主相关微生物组的泛化种和特化种在不同水产养殖系统中的分布。推测与自由生活的微生物组不同,宿主相关微生物群落加强了环境选择过程,以塑造共生微生物组,在多样化的异质环境下形成更多比例的泛化种。
中华鳖微生物组
生物学中最令人兴奋的进步是微生物组如何影响宿主的功能和行为。在所有生物系统中,宿主与微生物组构成共生功能体(holobiont),这种共生关系随着时间的推移共同进化。宿主遗传变异是否塑造了脊椎动物的宿主相关微生物组尚不清楚。大多数脊椎动物的肠道微生物群以厚壁菌门和拟杆菌门为主。鱼的肠道微生物群以变形菌门为主。我们的数据显示,中华鳖的肠道微生物群以变形菌门、拟杆菌门和厚壁菌门为主(图 1D 和表 S2)。海龟(Testudines、Reptilia)和夏威夷绿海龟(Chelonia mydas)的肠道微生物群以变形菌门、拟杆菌门和厚壁菌门为主。而幼年绿海龟的肠道微生物群则以拟杆菌门和厚壁菌门为主。在系统发育上中华鳖位于amniotic动物(鱼类和两栖动物)和amniotic动物(鸟类和哺乳动物)之间,在脊椎动物的进化中发挥着重要作用。这些结果表明宿主进化关系和肠道微生物结构之间存在联系。
动物皮肤被丰富多样的微生物组所覆盖。大多数皮肤微生物组研究都集中在人类、家畜和两栖动物身上。很少有研究探索水生动物、鸟类和鱼类的皮肤微生物组。非人类哺乳动物,包括牛、浣熊、松鼠、猪、狗和羊,以微球菌和葡萄球菌为主。鸟类的皮肤微生物组受地理位置、性别、饮食、圈养的影响。例如,笼养斑胸草雀的羽毛含有芽孢杆菌,被归类为“羽毛降解细菌”。清道夫新大陆秃鹰的皮肤微生物群以梭菌和梭杆菌为主。两栖类皮肤微生物组以酸杆菌门、放线菌门、拟杆菌门、蓝细菌门、厚壁菌门和变形杆菌门为主。鱼皮微生物群以变形菌门、厚壁菌门和放线菌门为主。中华鳖 (P. sinensis) 的皮肤微生物组以变形菌门、拟杆菌门和放线菌门以及不动杆菌属、气单胞菌属和罗氏菌属为主(图 1D 和表 S3)。有必要探索各种动物的皮肤微生物组,以了解动物如何与其微生物组共同进化。
RFC 中华鳖肠道中富集发酵多糖的微生物和益生菌
许多肠道微生物拥有多种酶来帮助宿主水解植物多糖。我们观察到,与IPC相比,在RFC的肠道微生物组中,富集更多的厚壁菌门和拟杆菌门、毛螺菌科、瘤胃球菌属、梭状芽孢杆菌属、罗斯氏菌属、拟杆菌属、普氏菌属、双歧杆菌属、真杆菌属、粪杆菌属和乳酸杆菌(图 3A 和 S4)。这些物种都与多糖发酵有关,表明 RFC中华鳖肠道微生物群的多糖发酵功能高于IPC。厚壁菌门在食草脊椎动物的粪便和肠道微生物群中占主导地位,在多糖发酵中发挥重要作用。纤维素分解细菌,如瘤胃球菌和罗斯氏菌,能够消化多糖,从膳食纤维中获取纤维素原纤维,通过代谢互补,其他成员可以间接利用这些原纤维。其他成员可以使用可溶性增溶寡糖和多糖,例如产生丁酸盐的丁酸弧菌、罗斯氏菌和梭菌,产琥珀酸的拟杆菌和普氏菌,产乙酸的梭菌。此外,毛螺菌科、真杆菌属、粪杆菌属、厚壁菌门的乳杆菌属和放线菌门的双歧杆菌属可以将多糖降解为短链挥发性脂肪酸(SCFAs),是草食动物肠道和粪便的特征。有许多关于脊椎动物SCFA 与饮食、健康、肥胖和代谢疾病之间关系的报道,特别是在人类和模型动物中。饮食对中华鳖的影响需要更深入的研究。然而,很明显,RFC中中华鳖的重量和体型明显小于IPC(图 7A 和表 S1)。人们普遍认为,食用富含脂肪的饮食的宿主可能会增加变形杆菌和梭状芽胞杆菌的相对丰度。与RFC相比,IPC的肠道微生物组中变形菌门的相对丰度要丰富得多(图 3A)。总之,我们的研究结果表明,RFC 中的中华鳖肠道由更多比例的发酵植物细胞壁多糖产生SCFA的微生物组成,从而促进能量代谢和健康。相比之下,IPC中的中华鳖肠道由更多比例的具有发酵脂肪能力的微生物组组成。所以,我们观察到RFC和IPC之间的肠道中存在不同浓度的脂肪酸和结合物、单糖(图 6 和表 S10)。此外,我们观察到脂肪滴在RFC中华鳖肝脏中显著积累(图 7B,C)。这些结果表明,两种水产养殖系统的中华鳖的脂质代谢和能量稳态存在显著差异。
乳酸杆菌和双歧杆菌是最广泛用于改善宿主健康的两种益生菌,它们与 SCFA、有机酸、维生素、胆汁盐、多酚、细菌素和甘油的代谢产生有关,并具有多种生物学功能。我们观察到,RFC的肠道微生物组中双歧杆菌和乳酸杆菌的相对丰度显著高于IPC(图 S4B)。总之,RFC可能通过招募更多比例的益生菌来有益于宿主健康。
代谢互补——宿主和微生物组之间的密切关联
微生物组和代谢组的整合分析被认为是评估宿主-微生物组相互作用的有效方法。我们发现两种养殖模式间肠道代谢物改变最大的的代谢途径涉及丙氨酸、天冬氨酸和谷氨酸代谢、GABA能突触、不饱和脂肪酸的生物合成、胰高血糖素信号通路、TCA循环以及精氨酸和脯氨酸代谢(图6C)和表 S11)。在IPC中通常投喂足够的饲料,中华鳖很容易获得足够的食物。然而,在RFC中喂食有限的食物,中华鳖更多的需要从周围环境中捕获食物,这可能导致IPC和RFC之间的中华鳖的饮食和活动模式完全不同。代谢物是宿主和微生物组相互作用的功能输出。因此,毫不奇怪地观察到两种不同的水产养殖模式对微生物组-宿主代谢和宿主适应性有很大影响,影响可能涉及能量稳态、脂质代谢、内分泌系统、抗炎活性等(表 S11)。事实上,在 RFC 中观察到中华鳖肝脏中脂滴(LDs)的含量显著增加(图 7B,C),这表明不同的水产养殖系统会影响中华鳖脂质代谢和能量稳态。因此,我们观察到龟体形状存在巨大差异(图 7A)。但需要进一步的研究来证实微生物组-宿主的相互作用。
肠道微生物生态学的一个主要挑战是确定影响宿主代谢过程和对环境变化反应的微生物成员。本研究使用 MIMOSA 来量化每个微生物群对代谢物变异的贡献。我们鉴定出五种差异肠道代谢物(2-氧代戊二酸/α-酮戊二酸、N-乙酰基-D-甘露糖胺、顺式-4-羟基-D-脯氨酸、烟酰胺和L-丙氨酸)潜在的基因和微生物关键贡献者,分别是链霉菌科、红环菌科和消化链球菌科、诺卡氏菌属和鲸蜡杆菌属(表 S13 和图 6D,E)。值得注意的是,这些物种并非都是变化最大和最丰富的微生物群(表 S13)。此外,每个预测良好的代谢物都有多个导致代谢差异的关键物种。例如,粘球菌目、链霉菌科和红环菌科、诺卡氏菌属的 ASVs 促进了顺式-4-羟基-D-脯氨酸的降解,Beta变形菌门、消化链球菌科、瘤胃球菌科、气单胞菌科的 ASVs,毛螺菌科、拟杆菌属和假单胞菌属促进了 2-酮戊二酸的合成(图 6E 和表 S13,14)。同时,一些关键物种是多种代谢物的主要贡献者。例如,消化链球菌科是 N-乙酰-D-甘露糖胺和烟酰胺的主要贡献者。总而言之,关键贡献者分析表明,对代谢表型的潜在变化贡献最大的微生物群可能不是变化最大或最丰富的微生物,每个代谢转变都是多微生物组的产物。
总 结
水产养殖环境显著影响中华鳖的表型、微生物群落组装和肠道代谢物。微生物群落结构和构建对养殖模式的响应是组织特异性的,并部分通过肠道代谢物改变宿主的适应性。这种影响如何通过食物链传递到人类的营养摄入仍有待进一步探索。我们的研究结果为水产养殖系统对宿主相关微生物群落的组装、多样性和功能的影响提供了新的见解,并高亮了肠道微生物组-代谢组关联中差异代谢物的关键微生物和基因贡献者。理解水产养殖系统对宿主相关的微生物群落的影响将加强水产养殖实践的管理,以实现更可持续的蓝色食品。
引文格式:
Ding, Xia, Feng Jin, Jiawang Xu, Shulei Zhang, Dongxu Chen, Beijuan Hu, and Yijiang Hong. 2022. “The impact of aquaculture system on the microbiome and gut metabolome of juvenile Chinese softshell turtle (Pelodiscus sinensis).” iMeta e17. https://doi.org/10.1002/imt2.17
作者简介

丁霞(第一兼通讯作者)
● 南昌大学生命科学学院,教授,博士生导师
● 2008年博士毕业于浙江大学微生物学专业。在微生物学领域从事科研工作10余年,研究方向微生物-宿主(环境)互作。江西省杰出青年人才,中国青年五四奖章集体获得者。

洪一江(通讯作者)
● 南昌大学生命科学学院,二级教授,博士生导师
● 2004年获得中国科学院水生生物研究所博士学位。江西省水生动物资源与利用重点实验室主任。国家贝类产业技术体系,岗位科学家。井冈山绿色生态立体养殖综合技术集成与示范,首席科学家。国家公益性行业科研专项,首席科学家。江西省特种水产产业技术体系,首席科学家。主持国家重点研发计划“蓝色粮仓科技创新”重点专项,国家自然科学基金。中国青年五四奖章集体获得者。全国农牧渔业丰收奖二等奖。
更多推荐
(▼ 点击跳转)
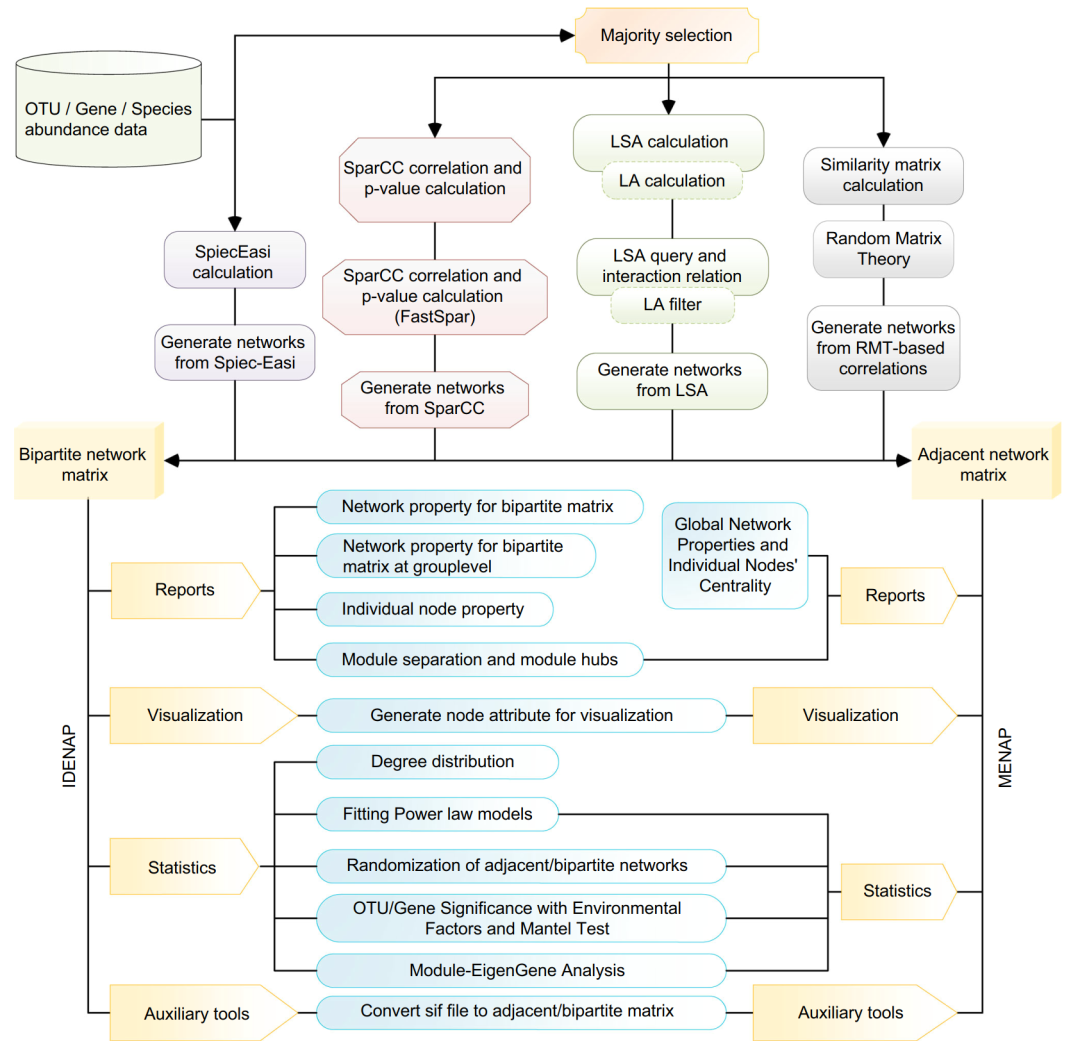
iMeta | 中科院生态中心邓晔组发布微生物组网络分析平台iNAP
iMeta | 华中科大宁康组综述用于蛋白质结构预测的宏基因组定量分析
iMeta | 中科院李小方等膳食甘草促进小鼠镉解毒并调节肠道菌群代谢
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析
iMeta | 南科大宋毅组综述逆境胁迫下植物向微生物组求救的遗传基础(附招聘)
iMeta:青岛大学苏晓泉组开发跨平台可交互的微生物组分析套件PMS

期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:[email protected]
微信公众号
iMeta
责任编辑
微微