之前写过两篇Excle数据转为plink的格式:
这里介绍一下常见的问题以及解决方法。
Excel格式的xls或者xlsx格式的文件
测序公司给的是xls或者xlsx格式的数据,数据的格式如下:
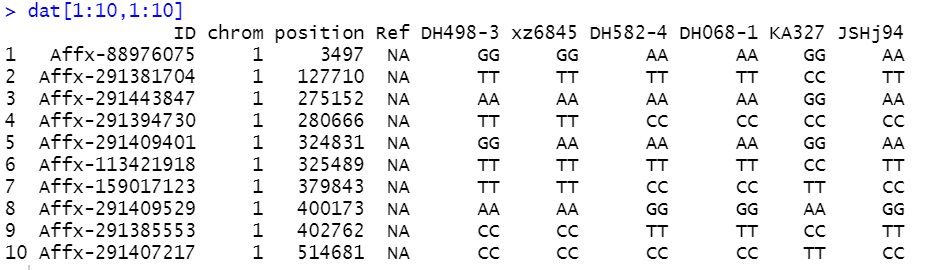
- 第一列是ID
- 第二列是染色体
- 第三列是物理位置
- 第四列是Ref
- 第五列以后是每个个体的具体分型,比如AT,AA,GG等分型。

这里,每一行是一个SNP,每一列是一个样本。
转化的代码
library(openxlsx)
library(tidyverse)
library(data.table)
dat = read.xlsx("genotype.xlsx")
dat[1:10,1:10]
map = dat %>% select(2,1,x = 3,p = 3)
head(map)
ped = dat %>% select(-c(1:4)) %>% t() %>% as.data.frame() %>%
mutate(ID = rownames(.)) %>%
mutate(x3=0,x4=0,x5=0,x6=0) %>%
select(FID=ID,IID=ID,x3,x4,x5,x6,everything())
ped[1:10,1:10]
fwrite(map, "file.map",col.names = F,quote = F,sep = " ")
fwrite(ped, "file.ped",col.names = F,quote = F,sep = " ",na = "00")
代码的逻辑:
第一,读取数据
第二,整理为map数据

第三,整理为ped数据

第四,保存为plink的格式
注意,这里的缺失定义为##,后面需要通过sed命令,将其转为00字符。

map数据:

ped数据:

使用plink命令判断是否转化成功
plink --file file --missing
如果没有报错,就转化成功了。
常见问题1:Error: Line 1 of .ped file has fewer tokens than expected.
这个一版是map和ped数据不匹配,可以通过R中的map和map查看一下什么情况:
> dim(map)
[1] 43251 4
> dim(ped)
[1] 185 43257
可以看到map有43251行,也就是有43251个SNP,ped比map多六列,因为第七列才是SNP的数据,结果没有什么问题。
再看一下map的前几行和后几行:

可以看到map最后几行是错误的,原始的xlsx文件有问题。
通过查看xlsx文件,发现最后有很多空白的内容,将相关行全部删除,再处理一下:


重新运行上面的代码:
$ plink --file file --missing
PLINK v1.90b6.21 64-bit (19 Oct 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to plink.log.
Options in effect:
--file file
--missing
32718 MB RAM detected; reserving 16359 MB for main workspace.
Possibly irregular .ped line. Restarting scan, assuming multichar alleles.
Rescanning .ped file... 0%
Error: Line 1 of .ped file has fewer tokens than expected.
还是报错。
常见问题2:缺失值为NN
这里,读取数据时,将其定义为缺失:
dat = read.xlsx("geno20.xlsx",na.strings = "NN")
再处理:
plink --file file --missing
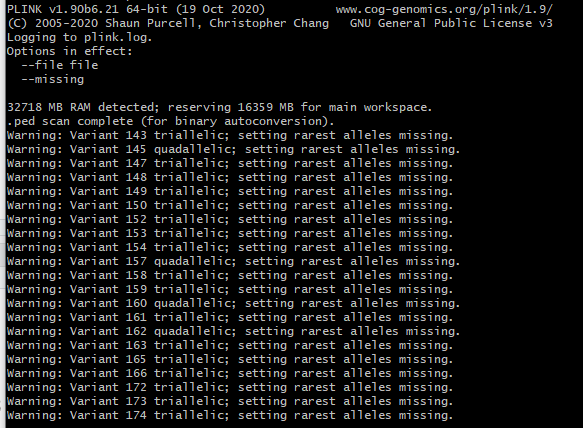
这样就读取成功了。
当然,上面的位点中,有些是多态性的位点,稀有的多态位点会作为缺失。
常见问题3:indel位点
plink格式不支持indel位点,需要将indel位点删除。
当然,如果有几万个snp,就不方便处理了。
思路:
- 将其读取到R中
- 转置
- 保存到本地
- 然后通过grep,去掉相关的行
- 然后再读到R中,再进行处理。
报错总结
- 数据有空行,有缺失,有indel。更新的代码中,判断是否有空行,将NN作为缺失读取到R中,可以避免上面的情况,更新后的代码如下:
library(openxlsx)
library(tidyverse)
library(data.table)
# 将缺失的分型定义为NN
dat = read.xlsx("genotype.xlsx",na.strings = "NN")
dat[1:10,1:30]
# 检查map是否正常
map = dat %>% select(2,1,x = 3,p = 3)
head(map)
tail(map)
ped = dat %>% select(-c(1:4)) %>% t() %>% as.data.frame() %>%
mutate(ID = rownames(.)) %>%
mutate(x3=0,x4=0,x5=0,x6=0) %>%
select(FID=ID,IID=ID,x3,x4,x5,x6,everything())
ped[1:10,1:30]
fwrite(map, "file.map",col.names = F,quote = F,sep = " ")
fwrite(ped, "file.ped",col.names = F,quote = F,sep = " ",na = "00")