compartment、TAD处理完之后,就是处理loop了;
实际上本人建议的处理顺序是compartment、loop再TAD,因为TAD分析过程中误差较大;
当然实际分析中TAD以及loop比较有价值。
其实这么一路分析下来,可以发现不需要花费那么多时间分析3个模块,直接使用juicer未尝不快,当然当初执行项目的时候实际上是从24年2月底到4月上旬左右,勉勉强强算是1个月半。当时就有人调侃,undergraduate实际上只需要12个月就能做完design,所以真这么想的话,其实走复现这一条路,如果获取了scripts,当初应该能够将时间缩短在1个月内,也就说当时是能够直接在23年910月左右就做完的。想想看后面所浪费的时间,只能说个人的潜力是无限的,只要精进一次,那么做经验中的事情速度只会更快。
#1,识别loop:
主要使用工具mustache,也就是区室分析中dchic的同一个lab
对照官网指示https://github.com/ay-lab/mustache
(1)首先是dchic的使用,参考https://github.com/ay-lab/mustache,用于识别loop
参考脚本https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/loop_call.sh,
即HiC_pipeline/pipeline/hic 基础pipeline骨架/01_loop_call.sh,
直接使用mustache.py函数

输入数据可以使用hic或者是mcool,以下挑选mcool文件为例,


其他参数都没有问题,主要是

参考了很多mcool或者是-norm的issue,
https://github.com/ay-lab/mustache/issues/45
实际上这里的bias value等同于矫正因子,所以直接使用mcool的默认即可(就是weight列)
事实上还是矫正的问题,可以参考我前面的hic数据格式转换部分的博客:https://blog.csdn.net/weixin_62528784/article/details/142106516?spm=1001.2014.3001.5502,

https://github.com/deeptools/HiCExplorer/issues/445
 总之cooler balance不够用不靠谱,建议是hicexplorer 2件套!
总之cooler balance不够用不靠谱,建议是hicexplorer 2件套!
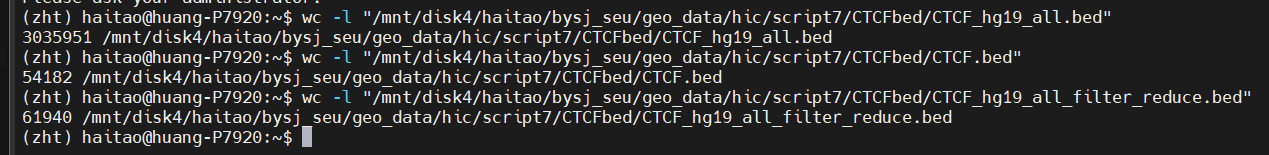
还是那句话,要知道**我们的hic-pro转换到cool,先是hicNormalize --normalize smallest再是hicCorrectMatrix --correctionMethod KR,然后合并为mcool,总之mcool文件经过了KR矫正!”**
问题很简单,
**什么数据格式用什么矫正方法(KR还是ICE)?
矫正之后的矫正因子在哪一列中(weight还是KR列)?**
我们用的是KR矫正,然后矫正因子列在mcool文件的weight列
而.hic数据格式是因为本身矫正之后因子列在KR中,所以直接处理hic格式是对KR列,
在hicexplorer的转换函数中如果要转换为cool格式可以设置correction_name为KR,然后出来的cool格式中矫正因子在weight列,那就是上面一样,使用mcool的weight列即可
总之,这是个英文名词学的问题,要多积累hic数据处理中的名词 correction factor!
https://github.com/deeptools/HiCExplorer/issues/681


或者直接去问开发者https://github.com/ay-lab/mustache/issues/66
但是因为我们使用的是mcool文件,实际处理过程中我们暂时没法合并,只想要指定10kb左右的数据,所以mcool要指定其中的res数据;
另外有个问题,很多issue中提到了mcool以及hic格式在mustache中会产生不一致数目的loop
https://github.com/ay-lab/mustache/issues/20
建议先试用juicebox对自己的结果进行一个判断分析,
整体上据说是hic格式更可靠一点;
那就从上游hic-pro——》juicer脚本转换为hic——》再然后看看需不需要在juicer或者hicexplorer中矫正一下——》在然后对接这里的mustache
(2)参考脚本https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/00_preprocessing.Rmd,
原始脚本为PDXHiC_supplemental/00_preprocessing.Rmd,
最后整理为HiC_pipeline/pipeline/hic 基础pipeline骨架/00_preprocessing.Rmd,
注意此处:
00脚本能够接受多种数据的输入input

使用mustache_results_v2,得到HMEC vs BT549对应的res下的tsv文件,比较完善(bed)
也有使用HiCcompare_results_v2,得到同上相应分辨率下的bedpe文件
甚至可以使用NeoLoopFinder,对应分辨率下的txt文件
也有使用GENOVA处理,得到pairedBED文件
还有SpectralTAD
以及hicFindTADs
此处统一使用mustache用于识别loop数据。原始脚本中此处可以导入更多数据格式,TAD以及loop。
然后实际上在处理样本过程中有个合并与否的问题,这里其实是一个分析的一致性的问题,因为上游分析过程中有些compartment分析、TAD分析软件有的能够合并,有的却不能够合并,所以会有不一致;
样本合并的问题:
分析mustache输出loop的tsv文件格式,两个anchor
①anchor的bed数据合并,就是从基因组位点上进行bedtools intersect等合并,但是loop tsv文件其他列的数据如何修改,对其取均值?
②另外一种策略就是loop合并比较麻烦,要anchor两边交集,可以保留loop数据,下游分析取出单边的anchor数据然后进行交集合并等
本项目因为数据量的问题,1vs3,暂时保留TNBC 3类loop数据,不合并为TNBC;
所有处理都是TNBC中1类vsHMEC,即3个1vs1的。
后期的1vs3处理方式不是从00脚本中的bed文件开始处理,而是直接从上游最开始mustache call loop得到的tsv文件(也就是loop的最原始文件)开始,
依旧参考00_preprocessing.Rmd



(3)分析识别common共有以及unique/specific特异的loop以及anchor:
见脚本00_preprocessing.Rmd;

只要两个anchor位点有交集就作为common,否则为unique;
1个loop2个anchor中只要有一个unique的anchor那就是unqiue的loop,两个都是common的anchor的才是common的loop。
另外对其loop以及anchor的统计信息:
创建条形图等
原始脚本PDXHiC_supplemental/Fig6_HiC_Loops/01_Anchor_Loop_Barplots.Rmd,
参考https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/01_Anchor_Loop_Barplots.Rmd,
整理为HiC_pipeline/pipeline/hic 基础pipeline骨架/01_Anchor_Loop_Barplots.Rmd

然后在原始脚本在处理loop数据的时候,先是按照anchor来处理的(先将loop拆分出来2个anchor,然后分析common以及unique),之后再按照loop分析common以及unique,
**需要注意两个问题:尤其是reduce,涉及到下游分析的可靠性
loop以及anchor的数据拆分出来又如何还原对应等?
anchor数据中的reduce问题(即overlap区域合并问题)?**
然后对loop以及anchor的size的统计见:
原始脚本见PDXHiC_supplemental/Fig6_HiC_Loops/03_Loop_Width.sh
整理见HiC_pipeline/pipeline/hic 基础pipeline骨架/Loop_Width.sh
参考https://github.com/magmarshh/LoopWidth
#2,loop注释:
(1)gene注释:
对anchor注释到gene,
原始脚本PDXHiC_supplemental/Fig7_HiC_Enrichments/01_Anchor_Gene_Annotations.Rmd
整理https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/01_Anchor_Gene_Annotations.Rmd
参考见HiC_pipeline/pipeline/hic 基础pipeline骨架/02_Anchor_Gene_Annotations.Rmd,
核心在于使用ChIPpeakAnno::annotatePeakInBatch将anchor回帖到最近的gene上,当然chipseeker也可以做;


这个过程中可以合并不同res的数据,比如说res1 10kb,res2 20kb等等(原始脚本中处理方法,但是自己做的话不建议);

因为做的时候只有10kb,所以没有合并;
然后gene注释的话还是那个问题,也就是compartment部分博客中的问题,是使用gene body overlap还是TSS within;当然,这里除了直接使用peak注释的R包,也可以使用compartment中的区室部分,也可以出一个博客。
注释了gene,那就还是那个处理流程,gene作为对象就有很多方法以及模块可以做了;
比如说是功能富集分析:
loop的anchor注释到的gene的富集分析:主要是unique
注意此处是富集分析(超几何检验),而非GSEA

注意metascape中哪些模块要用,哪些不用
(2)SE以及TE注释:
识别SE以及TE,参考young lab的ROSE软件,后续可以使用CRCmapper,
如果官网使用的软件版本过低的话,可以到github中配置适合版本的软件(主要是依赖软件之类)
#此处待更新
总之,假设我们找到了所有的E,即allE,将其分为SE+TE,获取了对应的bed文件
(3)SE以及TE与gene的联合分析(EPC):
①HiC_pipeline/pipeline/hic 基础pipeline骨架/02_Anchor_Gene_Annotations.Rmd脚本此处有部分:
主要就是multi-hub,是从网络图论出发,寻找节点最多的SE或者gene,找hub节点

②单纯分析EPC互作,即bed层面上的SE-P-C或者是TE-P-C
因为我们做的是EPC,所以实际上要获取的是不仅仅是gene数据,而是启动子P数据,所以我们之前anchor注释的数据,即前面使用ChIPpeakAnno::annotatePeakInBatch将anchor回帖到最近的gene注释的,可以修改一下,获取启动子bed数据,
当然我们也可以直接获取启动子的bed文件,可以使用clusterprofiler包的相关函数,或者是使用gtf等注释文件自己去提取;
此处做EPC分析的基础,都是基于差异loop分析的结果,也就是1(3)分析的内容,
本人在差异loop的基础上,使用获取各样本的SE的bed文件以及启动子文件,当然我们依据前面unique的loop(也就是差异loop)的定义可以知道,我们的差异loop完全可以在某个anchor上overlap(即共有anchor),如果这个时候另外一端的两个diff的anchor正好共同与另外一个元件所overlap(跨越),实际上这样注释出来的EPC相互作用对就不是特异性的了,反而是共同的;
当然,这涉及到anchor的尺寸(通常在软件中是loop的分辨率,比如说是10kb)、以及通常顺式元件的size(其实后面我对所分析出来的SE做了个size统计基本上不会超过10kb、以及不同样本之间bed文件如何对应上,总之要分析overlap问题需要考虑周全;
当然也有1种方法,是我个人想的:
可以在各自的anchor上向外上下游延伸一定kb,然后之后分析注释元件只考虑落在anchor bed内,要全包,而不是只是部分交集,这样差异loop分析出来的就肯定是差异EPC互作对了;总之在实现上,有很多细节要考虑,以及算法细节等。
###下面以部分overlap的元件注释(不是全部within)为例:
loop注释,主要是两端anchor注释,狭义即EPC注释
思路上是:将loop拆分成左右anchor bed文件(注意要保留原来在同一行的loop id信息),然后分别进行注释,主要注释为启动子、TE/SE、other文件,增强子使用前面中识别的bed文件,启动子还是使用clusterprofiler开发者的工具获取的启动子bed文件(自定义TSS上下游边界,使用chipseeker获取promoter的bed文件)——》然后左右anchor带着各自的注释信息合并回来原来的bedpe的loop文件,当然我们知道一个anchor可能注释成很多元件(因为可能与很多元件相重合),所以左右anchor的bed注释数据必然会不一致,还原为loop时将带有同一loop id信息的anchor注释元件进行组合(anchor1:元件1,2,3等| anchor2:元件a,b,c等——》然后进行组合,将每个可能的注释组合都作为这个loop的注释,C31xC31等)。
编写了一个脚本,参考https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/sortLOOP/newEPC.sh,
见HiC_pipeline/pipeline/hic 基础pipeline骨架/03_EPC_bedtools.sh,做左右anchor的bed注释;
主要还是使用bedtools,熟悉各种参数。
接着是整合回来(同一loop的左右anchor整合在一起),
参考https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/sortLOOP/loopTNBC.py,以及相应的loopBT549/HCC70/MB231/TNBC.py脚本,
整理见HiC_pipeline/pipeline/hic 基础pipeline骨架/03_loopTNBC.py;
然后统计各个组合的比例以及数目(主要是TE-P-C以及SE-P-C为主),参考
https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/sortLOOP/statloop.py,
整理见HiC_pipeline/pipeline/hic 基础pipeline骨架/03_statloop.py;
类似的脚本还有参考了的(但是实际执行过程中并没有使用,作为废稿弃用,但是处理算法思想可以参考):
https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/findloop.sh以及
https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/find_functional_loops.py;
③然后获取了SE-P-C,其实就可以做一个功能富集分析:
这里也很奇怪,我是获取保留有gene信息的启动子bed文件,直接使用gene,
还是只用启动子文件没有gene注释,然后后面使用对应启动子的bed使用区室分析中的脚本以及函数将其注释到gene中;(后面应该使用的是此法)
当然也可以使用上面chip分析中的peak注释;
总而言之,这里的gene肯定能够和EPC中的启动子P对应上,
然后获取了gene就是做功能富集分析,这里的gene和前面的差异anchor注释的gene又不一样;
这里的gene我选取的是SE-P-C,
比如说TNBC的SE-P-C,就是TNBC中哪些gene受到了SE的调控互作;
HMEC的SE-P-C,就是HMEC中的同理,也可以理解为TNBC中没有受到SE调控的;
当然我们说了,前面分析EPC的时候不是很严谨,我们是基于差异loop分析差异SE-P-C,但是也有可能注释到同一SE-P-C上。
虽然SE没有明确的ID,但是筛选出来的gene是能够做分析;
可以分析哪些gene是在SE中获得了调控,哪些gene在TNBC中失去了SE的调控,也就是正反做差集运算。
后面的CRC其实也可以同样分析,但是有点难以解释CRC的意义?
**在差异loop的基础上去注释两端的anchor,但是unqiue的anchor注释出来不一定是不同的元件,这里其实涉及到anchor尺寸与元件尺寸的问题,
理论上来说一开始不应该分析差异loop,应该一开始注释anchor,然后根据anchor的注释来分析loop差异与否,也就是说,在差异loop分析上,我可以将没有overlap的loop注释为差异的loop,但是实际上很有可能这两个loop对应的anchor就在同一个EPC元件内,这里涉及到元件bed中id标识的问题;
所以可以去看一下juicer以及mustache本身diff是怎么处理得出的loop是不是真正差异的——》实际上1(3)中差异loop的定义是PDX nature文献中定义的,因为没有使用工具本身的diff loop功能,所以才会有下游差异分析差异定义的问题,也涉及到元件注释,不清楚juicer、mustache本身diff功能做的是什么,可以之后试试!!!**
(4)EPC其他注释方法:直接使用相关R包
实际上在00_preprocessing.Rmd脚本中也有部分代码使用用于处理EPC注释的:
只不过是使用了其他的R包

#可以再出1篇博客,如何找到能够解决自己问题的R包或者是python工具,直接去github搜索关键词+限制语言,或者是直接去bioconductor寻找R包
(5)EPC衍生分析:类似APA/ATA,从聚集分析角度去分析两个感兴趣区域聚集互作热图(即两个对象,两个bed之间的互作)
①APA分析:
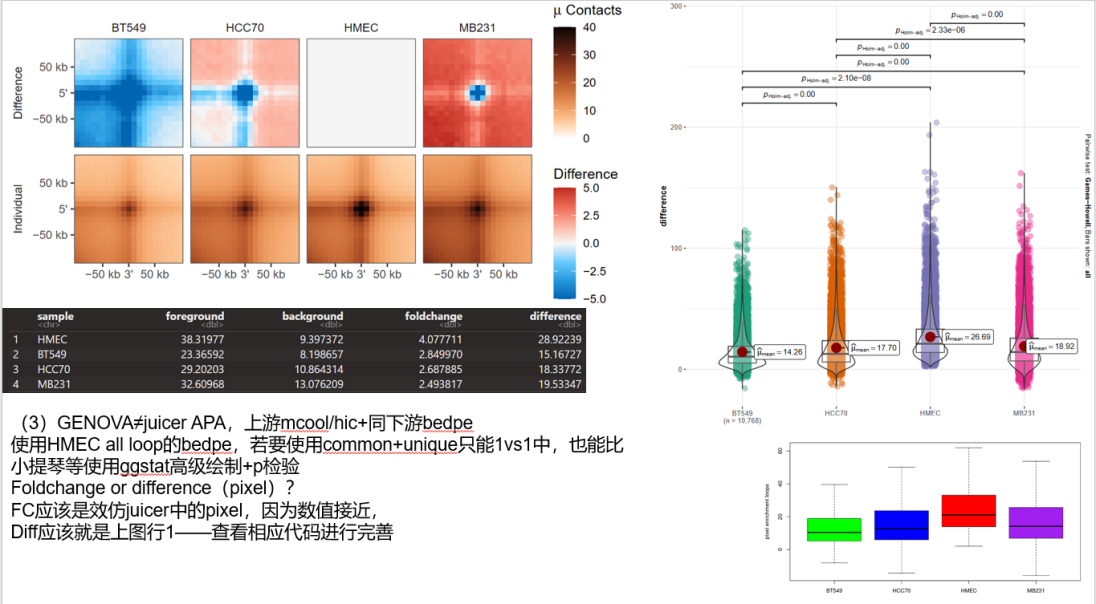

如果使用GENOVA的ATA函数的话,参考脚本:https://github.com/MaybeBio/TNBC-project/blob/main/2%2CCompartment/ABsaddle.Rmd,
整理为HiC_pipeline/pipeline/hic 基础pipeline骨架/04_loop_genova.Rmd,
实际上GENOVA部分所有loop的分析都在HiC_pipeline/pipeline/hic 基础pipeline骨架/04_loop_genova.Rmd中

对于APA的结果同样可以定量化quantify,然后可以利用中间数据二次发掘,做统计检验之类;

除了使用GENOVA的ATA函数做ATA分析,也可以使用juicer的ATA模块做,
原始脚本PDXHiC_supplemental/Fig6_HiC_Loops/02_APA_Mustache.qsub,
参考https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/APA.sh,
整理为HiC_pipeline/pipeline/hic 基础pipeline骨架/04_juicer_APA.sh,
实际分析过程中注意写好谁谁谁loop在谁谁谁中比较,即前缀后缀交叉记录号,比如说是TNBC特异性的loop在HMEC中不特异性等,交叉的文件都保留了下来,设置-u即call perchr的同理

注意这里的文件结果收集:
perchrAPA使用的是脚本中设置-u选项的,另外注意无论是交叉还是perhcr,都考虑跑完全部perchr+交叉后缀
因为perchr中最后也会有总结的全局的APA图

但是上面的图显示不完整,以及边界上的坐标和热图的色块bar不能够做到统一的像素max上限,以及四角的字体不能调节变大,还有图片上方的公式sub不能够完全显示,有一个P2LL值,就是因为显示不完整所以最后我裁剪修饰了,整理成上面的图样
所有的细节都看下面的文档:
Corner numbers correspond to center-to-corner ratios,另外如果在APA中设置-u的话可以处理perchr
https://github.com/aidenlab/juicer/wiki/APA
https://groups.google.com/g/3d-genomics/c/oFESb3oKVFA/m/I28BwyQnEAAJ
②PE-SCAn以及C-SCAn,具体参照GENOVA中的指南
实际上这里选取的就是各种bed文件之间的互作热图了,所以选取什么样的bed文件是重点;
可以是CTCF正反向的motif之类


#3,motif分析(即蛋白结合分析):
(1)首先是差异anchor或者是loop上的motif的分析,
因为实际上前面的分析中提供了很多的差异anchor、loop以及特异性SE、SE调控的gene等bed文件,都可以作为peak区域来分析motif等(当然范围仅限于启动子/gene以及增强子等);
当然在motif分析前应该是peak注释,对于bed文件,首选peak注释,再看motif等,当然实际上都是ATAC/ChIP的经典下游分析,所以实际上是同种分析模式;
依旧是04_loop_genova.Rmd:

实际上有了对loop的bed文件的注释,就很自然也会想到对TAD的bed做注释(同1个Rmd中)

peak注释好了之后,就是做motif分析了;
当然可以使用homer做分析,也可以使用R包做分析;
主要是使用meme包中的runAme函数,
参考https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/04a_Anchor_Masking.Rmd、以及https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/04b_JASPAR_MEME_IA.Rmd,
原始脚本PDXHiC_supplemental/Fig7_HiC_Enrichments/中对应04a以及04b,但是

这里又涉及到了ATAC数据区域的筛选问题,如果上游有ATAC数据可以使用,如果没有则只能使用数据库,原始文献就是使用了数据库;
当初执行时并没有选进行ATAC开放区域滤除,而是直接对差异loop(anchor)上的gene进行了motif分析,即04b脚本;
参考https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/04b_JASPAR_MEME_IA.Rmd,
整理为HiC_pipeline/pipeline/hic 基础pipeline骨架/06_JASPAR_MEME_IA.Rmd
(2)CTCF的motif分析:
实际上是参照了TAD以及loop两端的CTCF motif收敛或发散的现象;

首先是获取CTCF的motif的bed数据,当然如果上游有CTCF ChIP-seq数据可以直接处理分析,如果上游没有数据只能参考公共数据库了;
依旧是04_loop_genova.Rmd:

至于方向分析,参考https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/04_CTCF_Orientation.sh,
整理为HiC_pipeline/pipeline/hic 基础pipeline骨架/05_CTCF_Orientation.sh,其中对于CTCF motif方向的研究是https://github.com/magmarshh/CTCF_orientation,可以参考具体如何处理以及定义motif的方向,
原始脚本PDXHiC_supplemental/Fig6_HiC_Loops/04_CTCF_Orientation.sh;
要准备hg19等配套基因组的CTCF的motif的bed文件,见PDX开发者的R包CTCF,或者去motif分析数据库下载相应数据(meme,jaspar等);
还涉及到loop的motif问题,实际上jucier也有loop motif分析,但是执行效果不好,见脚本https://github.com/MaybeBio/TNBC-project/blob/main/4%2CLoop/juicer_loop_motif.sh,实际没有使用改脚本,待处理!
以下为mustache执行loop calling过程中的meme,可以跳过!!!!!!!!!
实际上在执行loop calling之后最重要的就是寻找diff loop,然后原始文献脚本中并没有使用mustache的diff loop功能,而是直接call loop,然后在R下游分析中对loop的bed文件进行了基础的anchor分析,以此来确定差异loop;
但是实际上mustache是有diff loop功能,能够直接找到差异的loop,所以这一步可以直接使用官方工具;



只有4个文件,类型上应该是tsv(总之都是bed文件格式)
所以要不要直接使用mustache中的diff功能做两两比对,还是直接做一个一个rep的tsv文件(就像原始脚本中),建议前者;
视脚本00需要,重新运行tsv文件获得步骤,
我感觉应该是能够直接依据新功能文件作为tsv的,但是为了兼容脚本中处理还是向下兼容文件处理;但是有点,diff是1.2.0就开发出来的,但是原文献中使用的是1.2.4,按理来说能用肯定可以用;反正不用直接使用diff功能,也能够直接识别diff以及用于下游分析。
然后当时在上游call loop的时候,实际上是只能处理1个样本1个样本,不知道如何将TNBC的3个生物学重复合并起来;

理论来说,依据上面call loop出来的结果,要想在上游合并TNBC 3合1的数据是困难的,
主要是call loop出来的结果的文件格式,即使是anchor的数据能够取交集,
但是除了anchor的另外几列的数据就很难合并处理了
当然如果后面对于loop的数据没有用上后面几列的话,实际上仅凭前面几列的数据如果也能行的话,实际上是能够直接在上游合并TNBC的loop的数据的;
①只要将TNBC的anchor的数据取交集,然后FDR等取mean等即可
如果后面几列的数据没有用上的话,实际上就是不影响的!!!
②或者是上游的loop数据依然保存,然后在下游分析的时候可以对取出的anchor等数据进行一个合并等
在处理完00_preprocessing.Rmd脚本之后,获得3对比对的loop以及anchor数据;


其实这里获得的结果也很明显了,
获得结果主要是对于anchor来分析,然后是loop,
分类是all,common,unique的数据
实际上对于loop而言:all的数据和原始call 出来的loop的数据是差不多是一样的;
现在来回溯:
在脚本00中处理的gi_HMEC等数据基本上就是HMEC all的loop的bedpe数据;

然后这两个数据都比原始的loop的数据要小


应该是这里去除了部分问题区域的数据,但又保证了最后的anchor的数据一致
重点在于如何识别common+unique的数据:
默认是当range任意部分与subject range有重叠,就被认为是overlap,这个默认type为"any"
实际上就是对findoverlap函数功能的认知

按照代码中的算法,第一个findoverlap实际上并没有找到common的loop,只不过是提供了一个common的大前提,
然后要寻找一致的loop的话需要深入到anchor角度上,要将anchor1vs1有any的overlap,2vs2有any的overlap,然后算作是有相同的anchor1+2,即相同的loop


但是有一很奇怪的现象:

从结果而言,最后获得所有的数据实际上是能够补全的
不对,上面的有问题,可能是3个的HMEC混合在了一起:
现在直接依据输出来判断所谓的overlap的效果如何:
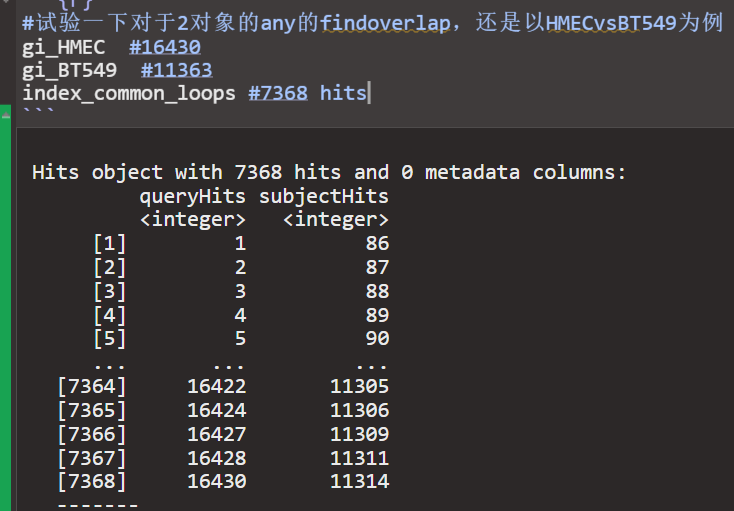
是最初的HMEC vs BT549的例子,
可以直接查看
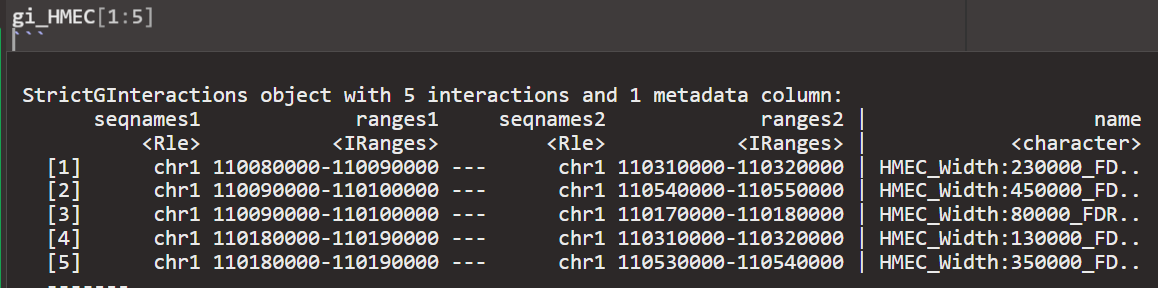

首先loop的数据中anchor2肯定比1大,因该是按照5-3’的数据来的
然后很明显的就是此处的数据基本上就是全部overlap的,然后每一个anchor的大小就是res数据的大小


其实看上面的这几行就能够很清楚的看到了所谓any的overlap,实际上鞍点1overlap的有,anchor2的也有,两个同时的也有
我感觉本身就有可能是因为call loop的算法奇怪,所以可能所谓的overlap会非常工整
比如说上面anchor1 overlap的,实际上anchor2的数据就已经是相邻的的
anchor2 overlap的,实际上anchor1的数据就已经是相邻的了
两个都overlap的,实际上就是equal了
所以这些数据非常神奇的,可能在大前提下找的any的overlap,真的就不可能是unique
所以该脚本中就有这个提示:

首先overlap的anchor,很明确可以算是同一个anchor,
但是相邻的anchor,就像上面那样,卡在边界的,理论上来讲应该先注释,再分析合并anchor,
因为res才10kb,anchor也就宽10kb,
很有可能有些相邻或者是不相邻的anchor,实际上是在同一个gene或者是enhancer内部,但是这些数据在初始分析的时候就因为单纯的分析被定义为了不同的loop
理论上来说一开始不应该分析差异loop,应该一开始注释anchor,然后根据anchor的注释来分析loop差异与否,因为统计上的相邻与否的差异不能表征生物学意义上的EPC元件的相邻与否
也就是说,在差异loop分析上,我可以将没有overlap的loop注释为差异的loop,但是实际上很有可能这两个loop对应的anchor就在同一个EPC元件内
所以都很难说的
理论上来说我应该将HMEC与TNBC的loop的所有两端anchor都做一个注释,然后看两端注释的元件是不是同一个,这样定义为相同的loop,否则有差异就是diff
但是有些注释的话就会很麻烦,因为我目前只能注释E,P
那那些没有注释参考数据的loop,因为无法确定另一端的anchor的信息,全都是NA就很难判断
所以还是按照原始脚本的先diff分析,之后再注释
所以这里其实就有一个问题,要不要看一下其他软件对于diff loop的处理
比如说是juicer diffhicccups以及mustache本身的diff功能

所以实际上的相邻的鞍点的merge是对于common而言的,只要common思路对+操作对,从结果上而言最后的数目上确实是all-common=unique没问题。
loop、anchor数据的统计展示:按照脚本01
就是计数统计展示:画了loop以及anchor数目的

注意一下这里的anchor是如何分析的,如何统计的,为什么anchor不是loop数目的两倍
上面说的是loop数目的一个统计,然后这里对应的实际上是anchor数目的统计;
按照上面的操作以及对应的统计,loop数目实际上在操作前后基本上是没有丢失数据的,
但是anchor的数据分析的过程中使用了reduce+merge_adjacent
实际上的anchor的分析类似于loop的分析,但是相比loop的分析要简单一点,
先是直接将anchor12的数据合并在一起,然后依然是使用findoverlap函数找common,但是相比较于loop,这里的common的anchor能够直接使用,
然后就是差集取unique数据,
然后就是reduce,这一步其实就是合并anchor范围数据
下面其实就是reduce前后的anchor数据:


其实对比是比较明显的,就是左边相邻的anchor,在reduce之后就会合并到一起,所以数目上几乎是下降了一半
但是这里实际上处理的同一个loop对象中,所以可以说是比如说是HMEC中某一个loop的某一个anchor以及另外一个loop的某一个anchor,因为anchor区域的相邻所以就使用reduce合并在了一起,所以可以说是同一个anchor
因为在loop的层面,其实处理的是两个对象,是无法做到真正合并anchor数据的,但是在anchor层面其实就可以做到;然后合并了之后其实是有实际bio意义的;
anchor后续还是先识别common,即使用findoverlap,然后再取差集获得unique的数据
另外一个问题就是anchor的数目问题:

注意到实际上脚本中anchor并没有reduce,所以有些相邻区域的anchor并没有合并,
但是因为这里的anchor实际上也没有用于后续分析,所以计数上也就没有计入
就很奇怪,前面reduce了,但是后面又还原过来了


那这个图能说明什么呢?
这里的common的anchor为什数目不一致,明确是reduce前的了,那么
实际上anchor的数据处理就是使用自己的处理分析方法,所以和前面的loop分析还是有点去区别的,当然也是取决于前面的loop分析的结果了,
然后loop分析的话实际上使用的方法除了上面脚本中使用的自己的方法之外,还是可以使用mustache中的diff的方法的,但是后续的anchor的数据处理以及分析的话,实际上就是只有目前的自己的脚本中的方法

可以看出:
实际上使用diff call出来的loop1,按理说应该是全部的loop in BT549
但是实际上一比对数目的话就会发现:
diff识别BT549全部11535loop,mustache是11364
HMEC是16642变成了16431

差异loop方面是:
diff识别的BT549的全部loop是11535,但是仅仅是使用mustche本身的call loop识别出来的数目是11364;
HMEC的话就是16642变成了16431,
diff的话是BT549 4153到3996
HMEC是9842到9063
可以说是自己脚本中处理出来的数目是比diff本身算法识别出来的还少

只能说自己脚本中识别出来的范围收束的会更快一点,跨度更大一点
loop的分析:
使用自己脚本,有了全部的loop数据——》diff的loop——》diff的anchor分析
使用diff分析出来的loop,本身也有全部的loop数目,也可以走上面那种分析1,
或者直接使用diff分析出来的loop——》那其实就直接省略了loop分析,可以直接走anchor分析了
暂时不适用mustache的diff分析,使用自己的分析
还有一个问题,实际上我自己在后续分析loop以及anchor数据的时候基本上没怎么用过FDR这个指标,按理来说和dchic一样应该在结果中再使用FDR筛选一遍
但是我最初在loop call中使用了这个
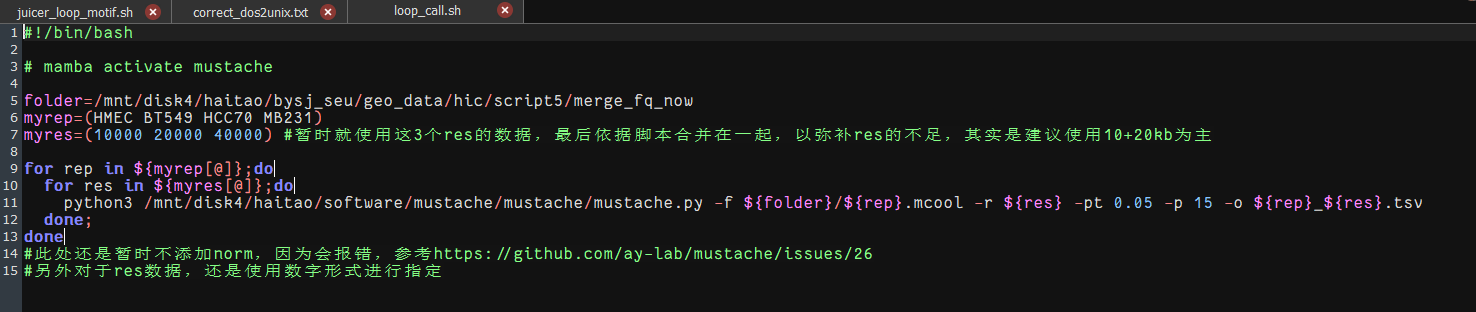
使用了0.05,应该得到了显著的loop互作,然后再有loop基础上进行diff分析,应该是没问题的。
后面分析APA,
要使用juicer tools,有juicer1.6最新版本https://github.com/aidenlab/juicer
另外有juicebox的最新版本2.20,以及从网站上拔下来的juicer_tools jar文件同版本
就使用后面这个
如果使用juicer_tools.jar 2.20,就要用java xx yy -jar命令(设置成2G,5G),最好能设置CPU模式
如果使用juicer_tools,那就不用使用java命令,但是会报错
https://github.com/aidenlab/juicer/issues/230,
根据上面提示
所以大概是要alisa将最新的juicer jar文件联系到jucier脚本上,所以juicer github中下载文件夹中并没有对应jar文件,就只能使用juicebox中更新的jar文件
之前的操作都是直接下载juicer的压缩包,然后直接解压再进入文件夹,所以使用的都是1.6版本的,并不是v2版本的;
但是如果使用git clone的话,据说是会使用最新的v2
但是其实文件夹中都没有对应的jar执行文件
但是下载下来的文件夹命名是不一样的


或者根据下面这个问题
https://github.com/aidenlab/Juicebox/issues/1028
对应的jar文件之类的都在juicebox仓库中寻找,然后再将juicer tools联系上去
那目前的操作就是要么直接使用juicebox仓库中的juicer tools jar文件,然后用java的命令格式
要么将juicer tools(无论是git clone还是wget),然后联系到上面这个jar文件中用alias

修改了一下,要么使用ln要么是使用alias,
但是都不太建议直接使用完整的文件名进行联系
算了,还是很麻烦,还是直接使用java -jar的格式算了
按照脚本中的情况来看,实际上和APA分析是有点区别的
就是使用的peak文件实际上已经拆分成了unique和common,而不是完整的total的loop的peak文件
(1)所以需不需要合并使用完整的peak文件,比如说用GENOVA或者还是juicer
(2)另外,注意到原始脚本中实际上是对HMEC,TNBC每个条件下都将所有对应的loop文件进行了APA分析,
即HMEC要对HMEC common,unique的loop分析,还要对应TNBC common unique也进行分析
同理TNBC还要对HMEC的也进行分析
我原本的计划是HMEC就对HMEC的,TNBC的就对TNBC的分析

当然有了这个文件,其实在GENOVA中也能够直接执行
总之,我自己HMEC vs HMEC,TNBC vs TNBC,
以及交叉的文件都保留了下来,设置-u即call perchr的同理

注意这里的文件结果收集:
perchrAPA使用的是脚本中设置-u选项的,另外注意无论是交叉还是perhcr,都考虑跑完全部perchr+交叉后缀
因为perchr中最后也会有总结的全局的APA图

但是上面的图显示不完整,以及边界上的坐标和热图的色块bar不能够做到统一的像素max上限,以及四角的字体不能调节变大,还有图片上方的公式sub不能够完全显示,有一个P2LL值,就是因为显示不完整所以最后我裁剪修饰了,整理成上面的图样
所有的细节都看下面的文档:
Corner numbers correspond to center-to-corner ratios,另外如果在APA中设置-u的话可以处理perchr
https://github.com/aidenlab/juicer/wiki/APA
https://groups.google.com/g/3d-genomics/c/oFESb3oKVFA/m/I28BwyQnEAAJ
loop宽度的分析:对照脚本03_Loop_Width.sh,主要是对00脚本中收集到的BT549/HMEC的common,unique的loop;
绘制了小提琴图以及饼图



以及相关统计量,TAD分析中也有相似的脚本
https://github.com/magmarshh/TADWidth?tab=readme-ov-file
除了使用PDX脚本中的CTCF python脚本之外,
还可以使用juicer中的工具
不对,这个是找loop中的CTCF的方向,不是为chip-seq CTCF peak找motif方向
但是暂时脚本跑不通!!!!!
后面分析CTCF motif时候需要准备hg19的CTCF的motif的bed文件:
(1)https://github.com/mdozmorov/HiC_data/blob/master/CTCF/README.md
主要是使用该文件:

awk '{print "chr"$2,$3,$4,$5,$6,$7,$8}' hg19.motifs.txt | grep -E "^chr(1?[0-9]|2[0-2]|X|Y)\b" > hg19_CTCF.bed

后来这个文件没有用上,这个文件实际上就是前面jucier中使用motifs函数功能时候所提供的公共数据,但是motifs那一步没有跑通,这个文件下面也有平替就暂时没有使用了
(2)按照这个开发者的CTCF的R包:
https://dozmorovlab.github.io/CTCF/index.html己构建motif文件
就是利用这个R的数据库自己提取数据库构建输出bed文件

基本上是仿造这里然后构建了hg19的在注释数据库中第一条的条例,
主要是一个原始的,一个是经过p值过滤的
最终脚本中使用的CTCF的motif的bed文件见下:

前两者见https://dozmorovlab.github.io/CTCF/index.html
使用hg19自己构建

第三个是在方向性脚本中获取的hg19的文件
https://github.com/magmarshh/CTCF_orientation/blob/main/test_data/CTCF.bed
注意里面的motif的bed文件都是按照方向性脚本&juicer motif功能中所提供的motif的bed文件的数据结构来构建的(之前跑出来大多是none或者是single其实也基本上是因为motif文件的结构不对)
所以里面的这些文件都是效仿正式的motif的文件结构进行了修正,
各自原始的文件在外面

从效果上来看,p值过滤的+CTCF脚本中提供的motif文件的效果差不多,毕竟文件大小差不多


没有p值过滤的效果就差很多了:

我总觉得应该用上真正peak的motif文件,所以上面数据库中预测的,无论p值有多显著,
实际上都不是真实的,但是不清楚如何获取对应真实的peak的motif文件
所以暂时peak文件用不上,只能用motif文件,暂时使用p值过滤之后的文件。
另外非常重要的一点是:
我需要搞清楚CTCF方向性文件中需要输入的bed文件是如何产生的,
前面直接寻找的是motif的bed文件,会直接有方向信息
但是自己产生的chip-seq的peak的bed文件,却没有方向性信息
所以重要的一点是,回想起来CTCF的方向,应该是从motif中获得的,但是我仅仅有CTCF的真正结合位点,然后该为位点信息是直接保留下来的,但是仅仅只是保留了坐标,并没有留下对应的序列的方向性信息
之前的文件:

所以这才是我用自己的peak的bed文件但是却始终获取不了方向的原因


narrowpeak其实没有给出那一条链,其实很正常,peak文件只能推断出CTCF结合的位点,但是实际的方向确实是motif推论的
所以我需要从peak的bed到motif的bed+方向信息(motif strand)
再仔细想想,需要自己的CTCF的peak数据吗,到底是如何处理的CTCF方向
只是筛选出CTCF peak吗,
真正的motif是不是还是需要到数据库中参考
那用公共的算了
https://github.com/liz-is/ctcf-motif-imr90/blob/master/imr90_ctcf_motif_direction.R



注意了,函数里说明了输入的motif的bed文件应该按照上面的方法进行整理
即列名都按照对应的方式来进行处理,当然了只需要几列:
主要是chrom,start,end,strand,其他几列都可以用NA填充

所以我这几个文件实际上都可以使用,只不过需要另外处理一下对应的几列
我觉得实际上可以直接提取出这3列,然后其他几列都用NA填充即可

就是用这几个motif文件作为公共。
开始处理ATAC以及ChIP的数据:
重新自己跑了一遍全局的代码,包括之前处理过的数据
然后在一个shell里面还是不能够使用mamba activate的命令



主要是trim galore中的输出文件夹需要提前建立
但是基本上bam文件是获得了
所以问题只有两个,一个是需要提前建立trim的输出文件夹,第二个是需要另外执行mamba activate 命令
至于执行脚本命令,可以使用&&,并且在输入命令的时候使用mamba执行哪个环境下执行哪个脚本
或者是直接将macs2安装在同一个环境下

目前是在rawfq文件夹中重新跑全程,主要是看qc报告
另外一个是在目前文件夹中跑macs2,主要是直接利用手头现有的bam文件,但是使用的上游fq文件是统一指定在3’端修建了2bp,但是不确定,所以要看报告QC



从效果上看大差不差都是3’ 2bp,所以第一次命令盲切是可行的
将目前手头上的bed文件都汇总在了一起

macs2:
https://github.com/macs3-project/MACS/blob/master/docs/callpeak.md
上面是对应输出的解释:
主要是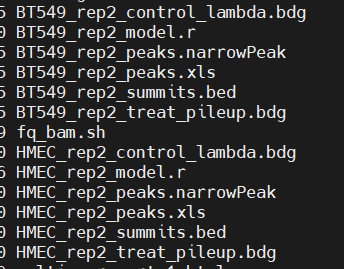
narrowpeak文件+xls文件+bed文件

官方说motif分析就是建议这个文件
但是实际上这个bed文件含有的是peak的summit即peak峰顶的bed文件,即很小的一部分坐标,即关于peak summit的一小部分的坐标,实际上不是常见的peak的结合的bed文件,
所以理论上来说也不应该使用这个peak文件,应该使用其他的文件
主要是参考了jucier tool中的motifs功能的界面之后,
https://colab.research.google.com/drive/1ucttsmbfJ7_HVw3VkWPSy-xqNKYF_VDh#scrollTo=qb-KAHrv5s2J一个juicer中的示例demo


下载了对应的peak文件之后,其实可以发现就是peak的完整坐标,而不是summit的坐标的,所以使用的就是narrowpeak文件
当然看样子在juicer中使用的bed文件只需要3列,就是chr+start+end
在script7中对应的文件夹里面建立
https://github.com/aidenlab/juicer/issues/227
主要是这里的说明不是很清楚,所以目前的情况是:
如果是1vs1的话,就是用同样的文件
如果是1vs3的话,就就将TNBC中3个peak文件做一个交集
因为我只有CTCF的peak文件,所以我本来想在inf中使用TNBC peak 3合1的文件,
un中使用对应1的peak文件
但是还是建议使用相同的文件:


理论上应该输入的是narrowpeak等文件,然后命名为bed文件
然后就是按照官方中的指示操作以及建立对应的bed文件,
对应的两个文件夹:
注意APA分析中使用的是jucier tools的v2版本,最新的2.2.20
然后motifs分析的时候会报错,所以还是使用旧版本的;
具体的CTCF的方向的分析在:
https://github.com/magmarshh/CTCF_orientation

需要考虑到两个anchor上哪条链上含有CTCF motif
其实并不是确定这种CTCF在EPC上研究有什么意义,
说不定我应该在TAD上试试看CTCF的方向性
可以逆向解析https://github.com/magmarshh/CTCF_orientation/blob/main/ctcf_orientation.py
脚本文件来分析如何获取对应CTCF 位点overlap的gene,
或者更切合实际的按照改脚本中所定义的方向分来来获取loop的坐标——
方向分类+以loop为单位获取坐标

后面分析特殊anchor的特殊loop:
(1)anchor intra内部比较:
比如说是SE内部区域,E内部区域,
首先收集srtp1中运行的SE结果文件


用的应该就是这两个table文件
暂时先将所有的SE文件都移动到script7中,相关的bed文件之后有需要再移动,以及有需要再重新rerun ROSE分析

===========另外基因组元件的bed文件:使用chipseeker获取promoter的bed文件
之后如果需要TSS或者是gene body的bed文件都可以使用
而启动子区域是没有明确定义的,需要自己指定,这里指定了上下游1kb
有个问题,就是C-Scan一般是用来分析
EPC,其他track之间的互作,
但是EPC的话需要提供P的bed文件,但是如上所述P需要自己定义
gene body问价我有,实际上能够从gtf文件中获取




后续继续GENOVA中的EPC研究:
首先是GENOVA中的PE-SCAn分析:
应该是用来分析远程的顺式互作,然后SE就是far-cis

PE-SCAn (27) creates virtual loop anchors by combining pairs of features within certain distance-thresholds and calculates the enrichment. CSCAn is an extension of PE-SCAn and allows multiple sets of peaks (e.g. enhancers and promoters or positively and negatively oriented CTCF motifs). It then creates virtual loops based on combinations of these sets. The discoveryobject of PE-SCAn and C-SCAn can be visualised and quantified in the same way as the APA, ARA and ATA.


the enrichment of interaction-frequency of all pairwise combinations of given regions
所以应该是将SE作为虚拟锚点,进行互作freq富集分析?
那应该是分析HMEC中的SE到TNBC中的变化,至少不会用上TNBC中的SE数据等?

https://github.com/robinweide/GENOVA/issues/297

PE-SCAn是提供一组区域一个bed文件,然后聚合组内区域之间的互作可视化,组内分析
C-SCAn是提供两组区域两个bed文件,然后是组间分析
这两个函数不局限于分析loop,能够将之前所有的3D基因组中的区域都提取出来进行分析!!!!!!!!!!!!!!!!!!!要全部及时跟进
如果要绘制intra,即一组区域,一个bed文件内部的互作富集分析,使用PESCAn:
一般是什么区域适合于分析内部intra的互作分析,可以提供的区域的bed文件有很多
首先是3D基因组的区域,可以是区室,TAD以及loop的bed区域
然后就是基因组调控元件区域,可以是promoter(或者是gene body+TSS位点等),然后就是E(可以是SE或者是all所有的E元件)
然后就是1D的多组学的track的peak的bed文件:这个可以是ATAC的开放性区域,或者是CHIP的组蛋白修饰+TF结合的区域的bed文件。
提供HMEC的SE的bed文件:

可以定量化之后再进行统计绘图分析+p检验
暂时没有找到区域绘制的需求,主要的功能实际上都由前面的ATA,APA等分析做完了,
按照后面主线需求补充intra模块
或者使用HICexploer进行聚合分析:
主要是https://hicexplorer.readthedocs.io/en/latest/content/tools/hicAggregateContacts.html
其他的工具都可以查看:

anchor inter互作比较,也就是提供两组区域,两个bed文件
可以使用EPC或者是SE-P-C,也可以使用motif方向相反的CTCF的bed文件
或者是使用多种1D多组学track
同样是可以使用
GENOVA中的CSCAn或者是hicexploer中的
https://hicexplorer.readthedocs.io/en/latest/content/tools/hicAggregateContacts.html
因为GENOVA中的EPC分析没有成功绘图,
所以暂时使用hicexplorer
脚本如下:
主要是绘制两个bed区域之间的互作富集的聚集分析,还在运行中!!!!!!!!!!!!

开始考虑如何合并TNBC中的loop以及anchor:
从最原始上游的tsv文件开始进行合并:
去除掉tsv文件后面,然后进行取交集的话


16642:929
数量上差距过大
而且这里直接进行合并的话实际上是没有考虑到loop的生物学意义,毕竟使用的区域仅仅只是两个res大小的anchor区域,但是实际上有些区域是联系在一起就是说是连续的,但是在仅仅对区域取交集的时候是没有考虑到这些区域的实际意义的,这些区域可能生物学应该取交集,但是实际上并没有取交集
所以后面不能仅仅从这些区域进行分析
所以放弃上游tsv取交集
但是00脚本处理之后


还是不行,虽然可以对这里的loop或者是anchor进行分析,
所以接下来的要么合并上游的tsv文件,
要么是3中取2的做法


最后的最后合并了,先将文件cat在一起不经过uniq,然后在R中处理



直接使用这个TNBC_10000.TSV
APA不能使用,因为没有TNBC的hic文件——GENOVA
但是juicer的motif倒是可以试一下。
开始分析SE:
与gene转录相联系,开始使用Fig7代码
将所有的enhancer以及SE的bed文件都转移进入了这里
仅仅保留了这几列,bed的3列+尺寸+是否是SE(1或0)



总之就是SE的分析中srtp部分分析的SE应该没有问题,但是最新的HCC70+BT549需要重新分析,可能需要重新分析

1,还是使用gene body,TSS的数据之后选择自己构建的promoter,或者看看原函数中对应的提供数据?
2,SE以及E的注释怎么处理?

运行anchor+gene的脚本01_Anchor_Gene_Annotations.Rmd
之后获得的gene:

主要都是一些freq的分析,类似于multi-hub之类

能不能做一个anchor位点的分析,就是分析anchor各自在什么位点
类似于anchor位点的注释,类似chip-seq的peak注释,理论上讲是可以类比chip等的分析的————分析anchor位点的

只做了overlap的,TSS的之后可以试一试
另外就是对于enhancer的注释,还没有试过,
所以enhancer注释试试,常规的chip的peak注释试试,以及TSS的gene注释试试
正式将enhancer与gene联系在一起,即进行EPC分析:
尝试使用脚本:https://github.com/kokonech/EPN_HiC_analysis/tree/main
直接从loopanalysis分析模块开始:

loop calling以及做过了,就直接从EPC开始:

修改了部分代码,然后可以直接使用对应的数据了
可以修改的地方如下:
-i是输入互作,指的就是loop文件,应该是bedpe文件,
查看原脚本中使用fithic:

或者使用在线网页工具:
https://www.csuligroup.com/EPIXplorer/Home
主要做富集分析的话就是对各自特异性的anchor上的gene做富集分析

文献上讲的也是对特异性的做GSEA,所以使用的就是HMEC以及TNBC中特异性的anchor的gene
但是使用metascape做不了,不知道为什么暂时出不了图,
可以考虑使用其他的在线网页工具等,或者是按照脚本中的GSEA来处理
1,再试试metascape等工具
2,实在不行就做脚本中的GSEA等
做metascape的时候可能gene太多了,超过了3k个,在TNBC中,可以试一下这些gene之间的交集,
https://metascape.freeforums.net/thread/40/more-3000-genes-analy
所以目前的做法,还是按照
1vs1 X3来做,然后gene可以取交集
1vs3,按照上面的说法就是合并之后分析
————————————
此处还是将1vs1 X3比对中的各种unqiue gene做一个交集
比如说HMEC内unqiue做交集
TNBC 3个内部做交集

1vs1 X3中获取的gene:各自unique取交集


3是取交集,32是3中取2及以上

metascape中HMEC取交集的,TNBC 2个都能上
然后使用这些分析结果
我将所有的E文件进行了排序:第一列是chr顺序,然后23列是bed文件顺序

还有前面的启动子文件
开始分析motif:


下载了左上中2的这个MEME

然后注意的是这里的04a的脚本没有执行,使用的是原始的loop以及anchor的数据,然后原始的04a脚本中所执行的操作是使用开放性等数据将loop以及anchor再筛选一遍,
大概就是筛选没有生物意义的问题区域,
但是那个脚本中使用的是开发者本人开发的一个开放性数据库,目前仅有hg38的数据,
相当于是没有ATAC的数据然后只用了hg38的公共数据,
但是我的是hg19,所以用不上,虽然我有ATAC的数据,但是操作一次太麻烦,而且没有必要
所以就直接使用原始的anchor数据了

还有一个问题就是anchor的reduce,
在获得原始数据的anchor,loop上没有使用reduce
可以试一试在最后的结果上进行reduce
这样以来后面的结果都要修改,脚本都要处理
暂时先不处理reduce的事情

之后全局回过头来考虑reduce的处理
https://snystrom.github.io/memes-manual/articles/integrative_analysis.html
在处理HMEC以及TNBC的问题上涉及到的motif数目太多,可能是前面加载motif数据库的时候有一个过滤条件:就是依据RNA-seq的表达数据进行筛选,只加载表达gene的motif,而什么是表达的gene,这个标准有待商榷,原始脚本以及上述文档中使用的都是》5,可能是原始的TPM值,可能是其他的数据表达数据,
但是我在处理的时候省略了这一步骤,所以获得motif数目太多(我是认为自己的RNA-seq的数据是在原来自己处理的脚本中已经筛选掉了一部分的低表达gene,所以在这里就没有再过滤操作了)!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!需要留心注意


filter the full motif database to select only those motifs corresponding to expressed gene.
自己的想法部分:
主要是multi-hub
SE对应的EPC:需要SE的bed文件
CRC如何
都可以参考hh那篇文献SE:拿到列表或者bed文件

对于SE部分:

原来处理的数据部分HMEC以及有了,MB231也用上了

现在手头上有一个脚本,用于寻找EPC的loop对:

但是涉及到一个双向选择的问题:
注意到是双向寻找,所以有一个参数threshold非常重要,使用的是2threshold的范围内寻找一个元件,


主要是这个函数:
看后面的使用情况,是用来给定了loop的anchor位置之后用于寻找附近的P以及E等元件,
比如说是给出左鞍点,如果寻找了P元件之后,那就再找右鞍点的E元件
然后因为后面的元件都是双向寻找,所以这个函数的功能实际上就是给定一个position,然后构建一个position±threshold的区域,即2threshold范围的区域,用于寻找对应的元件
其实仔细看这个脚本中的区域处理,应该是EP元件全部包含在anchor内部,
这个策略和前面我找EP的概念实际上是不一致的:
前面找E是overlap即可,不一定要within,
找P可以是gene body overlap,当然了这里提供的是P文件(实际上就是TSS文件),然后我设置的是上下游1kb,所以因该是P文件也是overlap(实际上就是TSS within)
但是这里的全部都是区域within,所以会严格很多,只能说策略不一样,
在R中是有函数findoverlap封装可以轻易完成任务,但是python中直接手撕手写函数功能就会比较麻烦
就当做是先试一下
获取的loop文件只有3列bed文件,看似是anchor的bed,不是looop的bedPE,
而且在处理中也确实是使用了position,所以确实是只有一个坐标数据,不是我认为的会将两个anchor的范围数据都做在一起,整合成一个gr对象
那么问题就来了,
如果要用3列的bed文件来表征loop文件,而且不是使用gr距离范围的的方法,应该如何表示,
实际上只能是获取两个anchor各自的summit的数据,然后分别作为loop鞍点数据中的左右anchor,
就相当于使用peak的summit的bed数据来表征peak的范围range
那问题就来了,
1,如何获取anchor的summit的bed文件,因为anchor的range都是10kb的,所以我可以依据现有的loop的数据,直接构造出新的loop的bed文件

给出一个bedPE文件,提取文件的1-6列
然后使用原始文件的第一列,第二列+5kb(5000),第五列+5kb(5000)


然后我获取了summit的bed文件,之后就是
2,考虑给出anchor的position数据之后,如何处理给定的E以及P的寻找范围的问题
如果是P的数据,主要是P的数据上下游1kb,所以全长是2kb,但是position实际上就是找左右的threshold的范围,然后如果是设计为5kb的话就是一个完整的anchor的数据了,当然也仅仅限制于还原原来的mustache中的数据,但是loop毕竟不一定是10kb长,
并且前面是要执行寻找within完全包含在里面的距离数据,所以得设置一个更加长的数据,其实我根据应该设置一个overlap会更较好,那样就没有必要考虑最后的距离的问题了
原来的计划中是anchor区域与P有overlap,
假设一个P是2kb,然后区范围是暂时设置为上下游5kb围绕summit,其实画一个图就可以看出来,应该设置为threshold 1kb到7kb(2kb+5kb),这样实际上就可以将原来计划中的anchor区域与全长的2kb的P区域的overlap的情况考虑进去,而且是极限情况,但是实际上具体涉及到的区域有多少个,就很难说了,会有很多个,
所以暂时设置为7kb大小,就定位threshold1
然后就是考虑enhancer的情况,这里实际上要考虑到具体的E以及SE的大小,可以通过查看对应的bed文件获知

其实光看尺寸的可以发现有很多的数据已经达到可几十kb的大小,
但是还是那句话,固然可以使用全部的尺寸数据,然后挑出一个最大的作为边界,但是还是算了
还是使用原始的ROSE软件中的设置,设置为12.5kb左右
那就17.5kb左右大小
如果是TE就会比较麻烦
所以还是仅仅试一下。
开始分析EPC:
loop的EPC互作分析:
(1)主要是对bedPE文件的注释以及分析:
主要是将loop中的左右anchor分开来按照顺序分别注释,然后将注释之后的数据合并在一起,注意顺序要还原,然后按照同一行中的loop的注释数据分别就进行anchor的分析,
主要是anchor主要是4类注释:
promoter,enhancer TE,SE,other等
其实bed注释之类的可以使用下面的各种bed工具进行分析

一个最原始的方法,就是使用bedtools做一个交集,
直接使用bedtools注释左边以及右边anchor:
我的想法就是如何不破坏loop的两段数据联系性,分别对anchor1以及2做bedtools intersect,分别使用TE,SE,P的数据文件做一个处理,最好在做交集的时候能够报告
anchor12都有哪些注释,如果都没有注释的,即NULL就作为other

启动子文件,TSS位点上下游1kb,但是没有gene的标注,虽然可以后期注释,但是建议前期使用的数据就是promoter,此处暂时先不使用P数据,然后后期获得注释的bed文件再分别注释gene定位
下面是TE以及SE的bed文件

主要是下面几个选项




用filename识别来源作为注释,loj或wao都可以保留
所以-wa±wb±loj=-wao,
-wa,-wb是为了完整输出record的原始参考信息,-loj是为了将没有overlap的也输出
还是用左边的代码-wa±wb±loj,还有一个是就是多个-b参考,比如说是TE,SE,P等,
如何得知哪些文件来源,
如果使用filename则会显示全部的文件全名,那样最后的文件会很长,而且每一行都不容易收集bed,
可以使用names,使用alias即别名,比如说按照顺序-b输入TE,SE,P的bed文件,那么之后我就可以直接-names TE,SE,P等

最后再加一个 -sortout,为了顺序上的美观
综上是-wa -wb -loj -names SE,TE,P按照指定顺序 -sortout
还有一个问题,就是
1,单个anchor会overlap很多区域,如何选,如果都保留,那么最坏一个loop会出现3
x3=9的组合情况
或者统一到一个区域,TE,SE,P都重叠就选SE的注释

见上面:注释到TE,SE,P的,注释为.的即为NULL
然后上面是直接使用loop的bedpe数据去注释,实际上是默认使用bed文件注释,也就相当于只注释了anchor1,可以在命令前面使用cut -f再标准输出
其实可以看到,因为一个anchor的尺寸是10kb,还是没有reduce之后的,
而SE以及TE,还有P的2kb都达不到这种程度,所以很有可能
一个anchor会注释到多个bed文件上去
按最后怎么分析

其实可以查看一下具体的分布:

发现还能到7:

所以是个问题?
2,如何确保anchor2注释顺序不乱


很显然,左边的anchor注释到的区域实际上更多
我在想,有没有一种方法,可以使用完整的loop的bedpe文件来连接两个注释好的bed文件(尽管不同行,但是能够将其按列合并在一起),
要么看R中如何处理,要们看bedtools有没有这种工具,merge之类
bedtools也有专门用于bedPE格式文件操作的工具,但是使用的时候没有办法接受多个bed文件的注释的输入,所以实际执行效果上就只能3个bed文件一个一个来注释,
但是这样处理的话就很麻烦,不能确保每一次注释之后的数据恩能够合并在一起
因为本来使用bed文件注释的话最后得到的3个bed注释的结果,就需要同时将两个bed的注释结果合并在一起,按照最后的loop的顺序
但是现在如果anchor注释上就将3个bed文件分开来注释的话,那么首先注释的时候会有两次的的anchor数据的对齐需求,
一次是对于单个anchor的数据需要将3个bed的注释对齐,
第二次是需要将两个anchor的数据按照loop中的顺序进行对齐
为了还原的需求,需要在原始的loop文件中设置一个loop id,然后做优anchor注释的时候也将对应的id提取出来,即组成bed3+loop id的格式
然后之后合并的时候就是按照左右anchor中得的loop id进行的一个还原合并
当然,可能会涉及到一个行数不一致的原因,那么到时候可以将所有注释都合并在一起,只提取注释列,
或者是不一致的行数就用空
暂时用使用获取左右anchor的代码获取了anchor带有loop id的bed文件,
但是有有两个文件是不一致的
就是说有两个文件有问题:主要是HCC70的左右anchor文件注释的获取


看报错应该是错误的-t制表符,然后再cut的时候出现了错误的切割
现在获取TNBC与HCC70的各自的loop的处理文件来进行比较,查看最后的分隔方式


其实实际上来看的话效果是差不多的,实际分隔的格式是一致的,
但是最后的效果不一样
那可能是对应的HCC70的SE以及TE的效果不一致

但是仔细查看之后就会发现实际上TE文件的格式是一致的,应该没有问题
所以看看SE文件的

发现确实是HCC70的SE文件有部分地方不一致

因为实际上的行数不是很多,所以可以直接在记事本中打开,然后修改一下效果的话其实是差不多的,就可以直接放在后面用了

然后现在就是获得了最后的左右anchor的bed文件+用于还原用的loop id,同时注意到左右anchor的文件上

首先我需要一个原始的连接文件,之后再记性多个注释合并的处理
最直观的方法,就是依据左右anchor文件的第4列,然后将两个文件直接合并,
如果左右两边哪个文件有多余的,另外一边就直接NA
或者能不能够直接组合分析
直接选取组合处理:
然后问题就是这里为了处理数据方便,直接提取注释之后的anchor数据的1-8列,即保留
anchor bed3列+loop id 1列+注释元件名称+bed 4列,一共是8列

新建了一个文件夹,重写脚本进行了分析,然后使用TNBC的loop的annotation的两个bed文件做分析

接下来要做的:

提取文件的第5,9列,然后做一个组合统计

主要绘制柱饼图+比率的雷达图,可以试试看矩阵热图。
(2)对基本的EPC进行了注释之后,之后就是将EPC提取出来,主要是提取SE/E-P关系
主要目的就是为了获取gene list:
提取各种E-P,SE-P,然后获取gene:
将各自unique的loop中E-P,SE-P都分析出来,主要是获取gene list,然后分析这个gene gain/lost E/SE,然后对gene做集合运算放到motif/gsea/建模上


3384符合,就用这个命令去提取


即提取TNBC unique中E-P以及SE-P的bed,有P的信息,可以进行注释以及gene获取
然后HMEC也同理获取一个gene list
然后进行分析比较

(实际上注释之后的loop数据只有左anchor+loop id+左右注释以及bed,因为py代码的原因没有补上右anchor的bed数据)
现在的问题就是获取P末尾3列进行gene的注释了:
需要动态提取P之后的3列



如果前期在P获取文件上带有gene名字的话,其实就可以直接获取gene list了,或者直接使用bedtools intersect gtf来获取相应的gene list:
但是还是使用下面loop分析中的代码来操作

测试一下效果:


这里实际上是对每个anchor注释到的P的gene,当然unique中的anchor会重合,所以P也会重合,所以实际最后注释出来的gene应该是小于这个数目
大致效果还是可以的:
另外因为P在代码中是reduce之后的了,所以实际效果middle不middle不影响
①现在手头上有了TNBC以及HMEC的unique的SE-P,TE-P
先分组:
HMEC中受TE,受SE,other
TNBC中受TE,受SE,other,还是9个组合
HMEC中受TE或other——TNBC中受SE
HMEC中other——TNBC中受TE,SE
要研究的是进一步的顺式调控,所以会比较麻烦,还是聚焦于SE上
能不能直接考虑TNBC中SE-PC - HMEC中SE-PC,
这样得到的差集无论如何都是TNBC中de novo的SE-PC
将效果集中在SE上,
真实调控关系如果一个gene受TE+SE,我会归类于SE,
这么来说,一个gene受TE/SE/Other调控,按照bio意义效果我都按照SE来,
那么我只需要TNBC中SE-PC - HMEC中SE-PC,各自做差集,
得到TNBC中de novo的SE-PC以及lose的SE-PC

原始列表在里面,然后写了个py程序做了个切出第一列,然后就是做gene的差集:


总是就是
282个de novo,51个lose,其他都是共有的
然后这一步就是对gene做一个GSEA就可以了
都放到metascape中:TNBC中de novo的SE-P的gene,lose SE-P的gene
TNBC本身全部SE-PC的gene
都做GSEA
然后最后就是取各种集合做一个模型:
其中TAD部分的gene不用考虑了
然后区室这一块的gene如果使用显著的gene的话,实际上就没有交集了

我实际上要的是BA+UP+SE那一块的gene:
15个实际上是可以进一步下降的,但是区室那一块太少了,
所以我决定使用全局的gene

但尴尬的是使用全局还是只有没有交集
要不要试一试全局XA,或者是显著的XA

但是看XA又没有意义,要不要依据这幅图看一看BA+BB:
先看看显著的

但奇怪的一点是,同样的显著区室的gene数据来做分析,最后在CNS中绘图效果是不一样的,所以最好还是使用CNS比较稳妥一点
然后还是这里的数据的问题
最后的集合就使用:
区室BA+de novo SE+UP的gene,至于说TAD,到时候就说是
转录放到了loop中,以及TAD的bio意义没有区室普遍,没有loop明确,所以没用
可以先画一个桑基图
做下游motif,或者是GSEA,建模等
可以分开来试一试
然后最后可以这么处理:
获取各自的SE-PC,然后对数据取一个差集
同时需要处理HMEC的unique以及TNBC的unique中EPC的变化


絮絮叨叨结束,以上可以跳过!!!!!!!!!!!!!!!!!!!!!!!!!!!
#4,其他loop可以做的:
(1)EPC等可以使用virtual_4c
virtual_4C,可以用于验证CRC之类的区域
暂时不知道该使用什么res下的数据,因为要涉及到loop之类的分析,暂时使用40kb数据至少
因为要使用的数据是一行的,所以不能使用全部的bed数据,建议使用CRC中的数据进行分析,建议使用SE中的CRC数据进行分析

可以将loop层面的转录调控分析引导到CRC上面去
下面是仅有的virtual4C的分析参考:
https://github.com/robinweide/GENOVA/issues/123
https://github.com/robinweide/GENOVA/issues/296

应该是visualize SE的viewpoint区域,然后添加SE联系的多个CRC区域,提供CRC的bed
(2)SE以及CRC,其实可以按照srtp进一步往下做,就是RTN寻找调控子,寻找特征gene,然后建模分析,生存分析
(3)GENOVA还有一些模块:
另外有个extend_loop的分析:

https://github.com/robinweide/GENOVA/issues/139


按照里面对于extend loop的分析实际上就是将所有的anchor数据提取出来,然后
组成一个任意anchor的组合,构成一个扩展的loop,当然包括已有的loop在里面

具体的分析可以到时候看对应的论文;
PS-SCAn、C-SCAn等,以及GENOVA本身就有一个关于SE的模块

实际上GENOVA中的工具分析的一般步骤:
①discovery处理数据:
常规使用的如下:

另外还有
 +绝缘评分分析+可以先visualize:
+绝缘评分分析+可以先visualize:
已完成:RCP,saddle,绝缘评分之后的tornado ,ATA,intra_inter_TAD
②进行定量化quantify:saddle
有如下对象可以quantify

—便于绘制统计p检验分布图:saddle+ggstat
已完成:saddle,ATA
③使用感兴趣区域bed进行交互可视化分析
—结合bed文件绘制tornado-plot

如上tornado聚合只能查看这3个对象
还有专门用于分析TAD边界上track信号的:

基本上GENOVA中的操作都是一样,先使用特殊函数进行discovery,
然后对该对象进行quantify,然后分析结果的数据结构,可以进行数据分析+绘图检验等
最后一并进行可视化分析+搭配不同track的bed文件等
最后可以仿照srtp中最后预测模型+CRC方面的内容进行分析
(4)CTCF可以深入:

对CTCF motif方向的深入分析等:
比如说正反向CTCF motif对的PS-SCAn、C-SCAn,
以及对https://github.com/dozmorovlab/CTCF_orientation其中的算法清楚,留下计算方向的子模块

(5)其他:



以及没有复现的PDXHiC_supplemental/Fig7_HiC_Enrichments/(04b不完全复现)

mustache的-norm问题——或者直接去问开发者https://github.com/ay-lab/mustache/issues/66
todo:
1,如何获取转录本、启动子等bed文件,从gtf等注释中
2,bed注释gene的方法
findoverlap、chip类peak注释函数,bedtools工具的使用等
3,每篇博客下面的todo
4,先占个坑
ChIP/ATAC:待更新
主要参考https://github.com/MaybeBio/TNBC-project/tree/main/5%2COther_omics,
https://github.com/mdozmorov/ChIP-seq_notes
主要就是参考seu-实践
以及srtp/投稿部分code(SE/CRC——》TCGA——》RTN——》cox model)套路走
5,hic数据处理pipeline
https://data.4dnucleome.org/resources/data-analysis/hi_c-processing-pipeline#recheck
其实很多问题在于:
1,如何找对工具,也就是一整个处理体系的工具:权威公众号固然可,但建议直接看CNS中的methods
2,之后如何设置正确参数集(还是建议看methods)
3,按照模块复现,一定要注释好,便于后面继承以及修改对接