- 数据详细信息介绍
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65168
从网站中下载编号为GSE65168 的数据集,平台是GPL6244【HuGene-1_0-st】,是Affymetrix公司的新一代芯片(WT),所以因此选择oligo包读取CEL数据,进行更进一步的处理。该芯片一共有八个样本(GSM1588481-GSM1588488),用cDNA芯片分析了正常/缺氧条件下VHL阴性786-O RCC细胞系和VHL转染物的RNA表达情况。从处理条件上,可以将样本分为两类(正常/缺氧);从是否转染VHL,也可以将样本分为两类(转染/未转染VHL)。
- 实验步骤
- 获取CEL数据。
从https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65168
网站上下载编号为GSE65168的原始数据集,在这里请注意,请下载.CEL类型的原始数据。
CEL文件应该是Supplementary file里的raw.tar,解压后就是所有样本的CEL文件。CEL文件是提交数据者提交的芯片原始数据,是Affmetrix公司的芯片格式,需要用专业软件如R打开,不可以被可视化。需要先对CEL文件进行质量控制和数据预处理(具体用bioconductor中对应的不同的包)。

而如果想直接进行分析,如差异基因筛选,可以下载Downloadfamily里的Seires Matrix Files,这是GEO工作人员将提交的原始数据进行整理和标准化后可以被可视化的txt文件,即每个探针的表达量。

下载原始数据压缩包,至本地文件路径(E:\大三下\0-转录组信息学\作业\ GSE65168_RAW)处。
第一步完成。
2.用R语言从本地路径下提取CEL类型文件。
打开Rstudio。
library(oligo) #加载oligo包
setwd("E:/大三下/0-转录组信息学/GSE65168_RAW") #设置工作路径
data.dir<-"E:/大三下/0-转录组信息学/GSE65168_RAW" #将CEL存放的路径存放在自定义变量data.dir处
(celfiles<-list.files(data.dir,"\\.gz$"))
data.raw<-read.celfiles(filenames=file.path(data.dir,celfiles)) #用oligo包中的read.celfiles函数提取CEL文件
data.raw

设置探针(样本)的名称。
treats<-strsplit("BR HBR VBR VHBR BR HBR VBR VHBR"," ")[[1]]
(snames<-paste(treats,rep(1:2,c(4,4)),sep=" "))
sampleNames(data.raw)<-snames
pData(data.raw)$index<-treats
sampleNames(data.raw)
[1] "BR 1" "HBR 1" "VBR 1" "VHBR 1" "BR 2" "HBR 2" "VBR 2" "VHBR 2"
表达矩阵
exprs_matrix<-data.raw@assayData$exprs
exprs_matrix[1:5,1:5]

3. 绘制MA图,查看各芯片中M,A之间的关系。
MA图可以很好的反映M和A两个变量之间的关系。
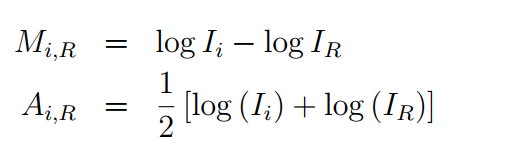
par(mfrow=c(2,2))
MAplot(data.raw[,1:4],pair=F)
MAplot(data.raw[,5:8],pair=F)


4. 运用oligo包的fitProbeLevelModel()函数,进行数据的预处理。
fit<-fitProbeLevelModel(data.raw)
#1) background subtraction
#2) normalization
#3) summarization
fitProbeLevelModel函数实现了芯片数据预处理的所有过程。

可视化fitProbeLevelModel结果
rle<-RLE(fit,type = "values")
boxplot(rle,col=rainbow(8),ylim=c(-1.2,1.2),main="RLE",ylab="RLE",cex.axis=0.7)
#各样本的众数在0 附近

nuse<-NUSE(fit,type = "values")
boxplot(nuse,col=rainbow(8),ylim=c(0.93,1.07),main="NUSE",ylab="NUSE",cex.axis=0.78)
#各样本的众数在1 附近

可见对芯片的预处理效果良好,芯片质量可靠。