转录组之序列剪切(Trimmomatic)
本文简介:这篇文章是经过了自己学习实践出来的,参考了很多资料,如若有大佬能指出错误,我将感激不敬,有错误,请在评论区留言,谢谢。 更新日期:2023年7月6号
本文知乎地址:https://zhuanlan.zhihu.com/p/642000061
在这里我们们先介绍以下为什么需要进行序列剪切;序列剪切(sequence trimming)是在测序数据分析中的一项预处理步骤,其目的是去除低质量的碱基和噪音,以提高数据的质量和准确性。
以下是进行序列剪切的一些常见原因:
- 去除低质量碱基:测序过程中,每个碱基都有一个关联的质量值,表示该碱基被正确测序的可信度。序列剪切可以去除开头或末尾质量较低的碱基,以提高数据的质量和可靠性。
- 去除适配器序列:在DNA测序过程中,通常会使用适配器序列将DNA片段连接到测序芯片上。适配器序列可能残留于测序数据中,对后续分析造成干扰。序列剪切可以帮助去除这些适配器序列。
- 去除重复序列:在某些实验中,可能会出现PCR扩增引起的重复序列,这些重复序列可能影响后续分析的准确性。序列剪切可以帮助去除这些重复性序列。
- 去除噪音和杂质:测序数据中可能存在噪音、测序错误或其他杂质。序列剪切可以帮助去除这些噪音和杂质,提高数据的准确性和可信度。
通过序列剪切,可以提高测序数据的质量和准确性,减少后续分析的误差,并帮助从原始测序数据中提取出更有意义的信息。
接头的由来
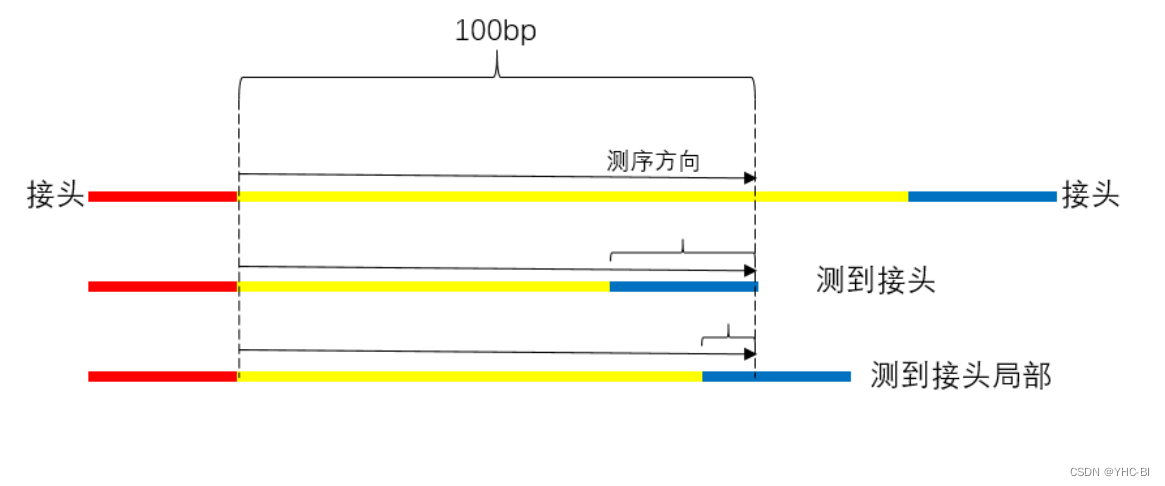
文库构建前,核酸经过随机打断,有的本身就长短不一(mRNA),因此接头之间片段长度也长短不一,而二代测序的测序长度一般是固定,肯定会有部分短于测序读长的序列被测序,因此测序序列中包含了部分或全部接头序列,需要进行接头序列的检测并过滤掉对应的reads或截掉接头序列。(如下图所示得蓝色接头部分,有些测到了接头(部分与全部都有可能),至于原因建议去看二代测序原理,这里内容过多,不宜展开)
Trimmomatic软件简介
Trimmomatic是一个广受欢迎的 Illumina 平台数据过滤工具。其他平台的数据例如 Iron torrent ,PGM 测序数据可以用 fastx_toolkit 、NGSQC toolkit 来过滤。支持多线程,处理数据速度快,主要用来去除 Illumina 平台的 Fastq 序列中的接头,并根据碱基质量值对 Fastq 进行修剪。软件有两种过滤模式,分别对应 SE 和 PE 测序数据,同时支持 gzip 和 bzip2 压缩文件。
上次我们使用FastQC软件得到质控后的结果,从结果我们可以得到,某数据是否需要切除序列,切除多少,是否有接头,在FastQC过表达部分可以看到有重复序列,重中发现相同的规律也可以使用该序列去掉不想要的部分(这句话当我没说过,因为我没试过)。
Trimmomatic软件的下载(只提供 Linux)
1、Trimmomatic软件的下载。
wget https://github.com/usadellab/Trimmomatic/files/5854859/Trimmomatic-0.39.zip
2、解压
unzip Trimmomatic-0.39.zip
配置环境(我配置在了~/.bashrc文件中 )

# trimmomatic
echo "export PATH="/home/cyh/biosoft/Trimmomatic-0.39: $PATH" " >> ~/.bashrc
3、测试 (help一下)
java -jar /home/cyh/biosoft/Trimmomatic-0.39/trimmomatic-0.39.jar -h
Trimmomatic软件的使用
官网参考地址:USADELLAB.org - Trimmomatic: A flexible read trimming tool for Illumina NGS data
部分参数简介:
ILLUMINACLIP: Cut adapter and other illumina-specific sequences from the read.
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 # 切除TruSeq3-PE中提供的Illumina适配器,去接头
SLIDINGWINDOW: Perform a sliding window trimming, cutting once the average quality within the window falls below a threshold.
SLIDINGWINDOW:4:15 #扫描4个碱基宽滑动窗口,当每个碱基的平均质量下降到15以下时进行剪切
LEADING: Cut bases off the start of a read, if below a threshold quality
LEADING:3 # 删除前低质量碱基(低于质量3)
TRAILING: Cut bases off the end of a read, if below a threshold quality
TRAILING:3 # 删除后低质量碱基(低于质量3)
CROP: Cut the read to a specified length
CROP:length #从reads开始开始所要保留的碱基数为length
HEADCROP: Cut the specified number of bases from the start of the read
HEADCROP:12 #删除前12个碱基
MINLEN: Drop the read if it is below a specified length
MINLEN:36 # 上述步骤完成后,删除小于36个碱基的reads (放最后)
TOPHRED33: Convert quality scores to Phred-33
-phred33 #表示将质量分数转换为 Phred-33
TOPHRED64: Convert quality scores to Phred-64
-phred64 #表示将质量分数转换为 Phred-64
“phred33"是一种常见的测序数据格式的表示方式,用于表示DNA测序中的质量值。在该格式中,每个碱基对应一个ASCII字符,表示测序质量的分数。其中,字符”!"表示质量值为0,而字符"I"表示质量值为40。这种表示方式主要用于Sanger测序和Illumina测序等技术中。
单端(例子):
java -jar trimmomatic-0.35.jar SE -phred33 input.fq.gz output.fq.gz ILLUMINACLIP:TruSeq3-SE:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
上述例子描述:SE表示单端,使用数据格式为33的;input.fq.gz表示输入文件,output.fq.gz表示输出文件,两个文件格式都为 .fq格式的压缩文件(Trimmomatic可以输入输出压缩文件);ILLUMINACLIP:TruSeq3-SE:2:30:10表示软件自带的Illumina适配器,用于去接头;LEADING:3表示前(端)切除质量低于3的碱基;TRAILING:3表示后(端)切除质量低于3的碱基;SLIDINGWINDOW:4:15表示扫描4个碱基宽滑动窗口,当每个碱基的平均质量下降到15以下时进行剪切;MINLEN:36表示删除小于36个碱基的reads(该参数需要在最后使用)
双端:
java -jar trimmomatic-0.35.jar PE -phred33 input_forward.fq.gz input_reverse.fq.gz output_forward_paired.fq.gz output_forward_unpaired.fq.gz output_reverse_paired.fq.gz output_reverse_unpaired.fq.gz ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
双端与单端的类似,其中指定PE参数,表示这是双端数据,因为是双端数据,所以输入数据有两个,并且这两个数据必须是配对的(即核酸链的正反两条数据),输出结果有四个,每条reads都会生成两个文件。 "output_forward_paired.fq.gz"表示通过trimmomatic处理后,前向(forward)读取序列中的配对序列(paired sequences)输出到该文件。 “output_forward_unpaired.fq.gz” 表示通过trimmomatic处理后,前向 (forward)读取序列中未配对的序列(unpaired sequences)输出到该文件。"output_reverse_paired.fq.gz"表示通过trimmomatic处理后,后向(forward)读取序列中的配对序列(paired sequences)输出到该文件。 “output_reverse_unpaired.fq.gz” 表示通过trimmomatic处理后,后向 (forward)读取序列中未配对的序列(unpaired sequences)输出到该文件。在指定接头适配器时注意时PE(ILLUMINACLIP:TruSeq3-PE.fa:2:30:10)
HEADCROP:12 表示删除前12个碱基
注意:如接头文件不在当前目录下,要指定,写全路径,否则报错。
实际举例:
在这里我使用了一对(正反数据)SRR12415652_1.fastq与SRR12415652_2.fastq,这个数据在数据库能下(至于时NCBI还以TCGA因年代久远,搞忘了)
java -jar /home/cyh/biosoft/Trimmomatic-0.39/trimmomatic-0.39.jar PE /home/cyh/Desktop/fastq_dir/SRR12415652_1.fastq /home/cyh/Desktop/fastq_dir/SRR12415652_2.fastq /home/cyh/Desktop/SRR124_result/SRR12415652_1_paired.fq.gz /home/cyh/Desktop/SRR124_result/SRR12415652_1_unpaired.fq.gz /home/cyh/Desktop/SRR124_result/SRR12415652_2_paired.fq.gz /home/cyh/Desktop/SRR124_result/SRR12415652_2_unpaired.fq.gz ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 HEADCROP:12 MINLEN:36
跑完数据后,我们需要对生成得结果(只需要paired的)进行质控(FastQC),再一次看一下质控报告。
结果如下:(SRR12415652_1的,2 的差不多,不展示了)
-
Per base sequence quality部分
剪切前后对比
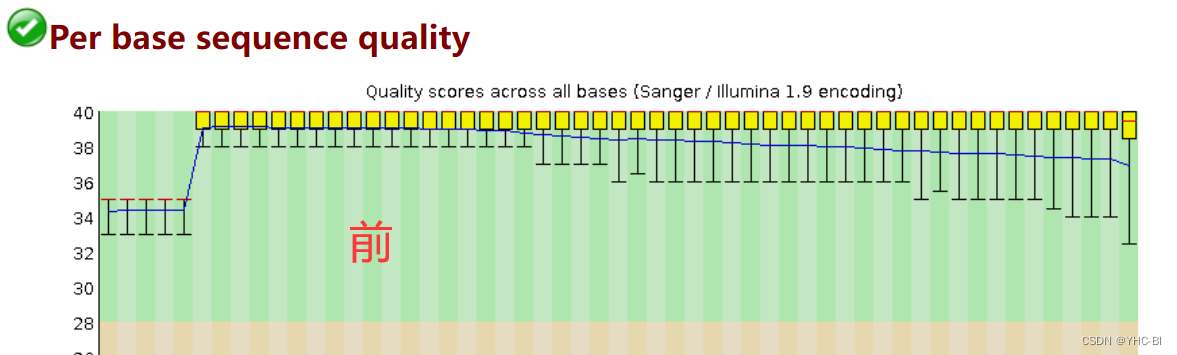
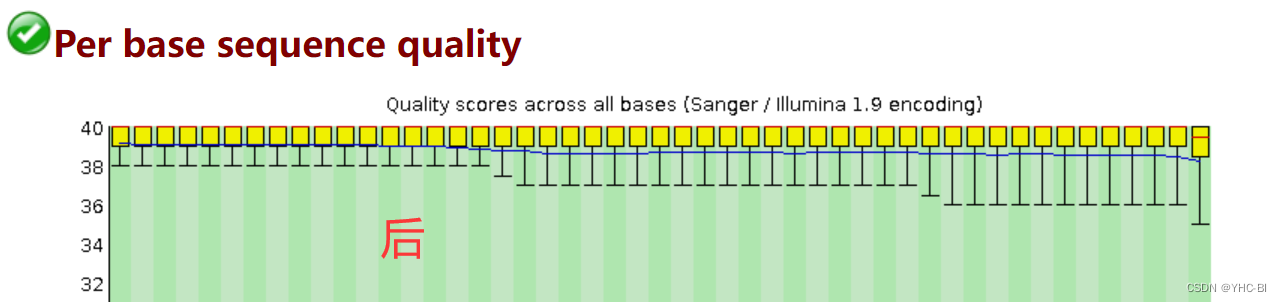
这部分肉眼可见的完美,
-
Per base sequence content部分
剪切前

剪切后
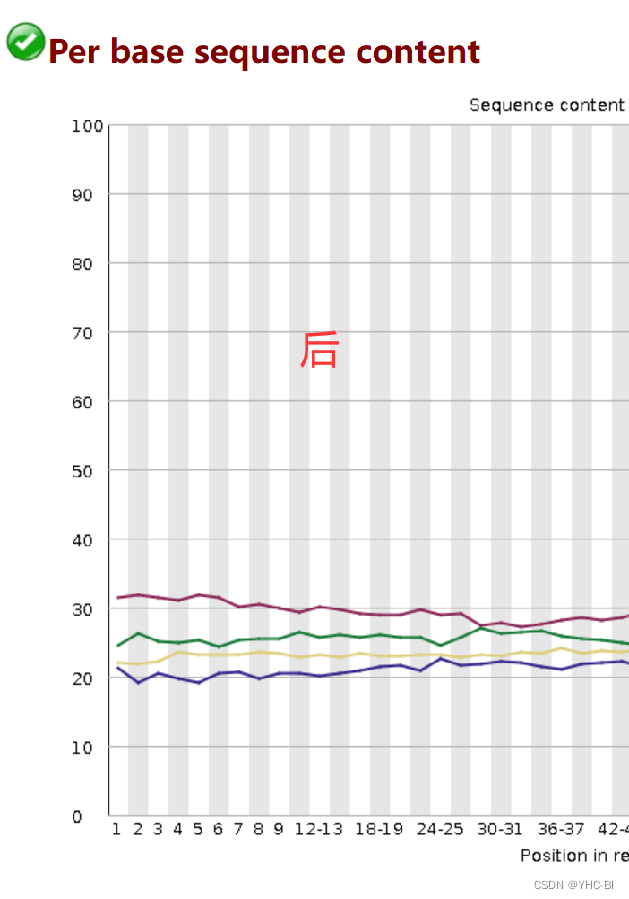
这部分我因为删除了前12个碱基,所以前面就没有了。(这个其实可以不切,看需求吧)
-
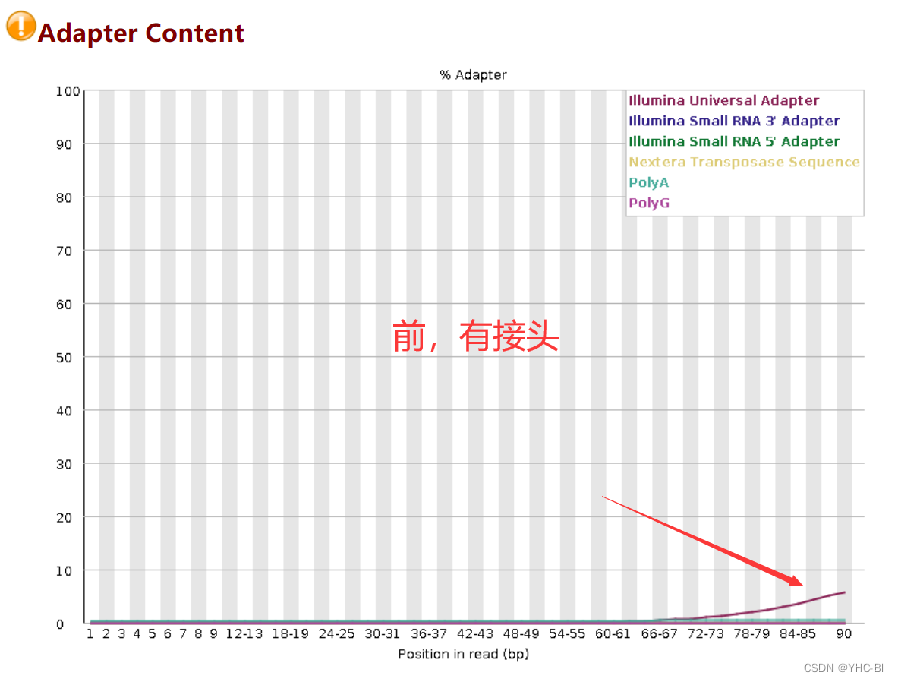
Adapter Content 部分
剪切前
剪切后
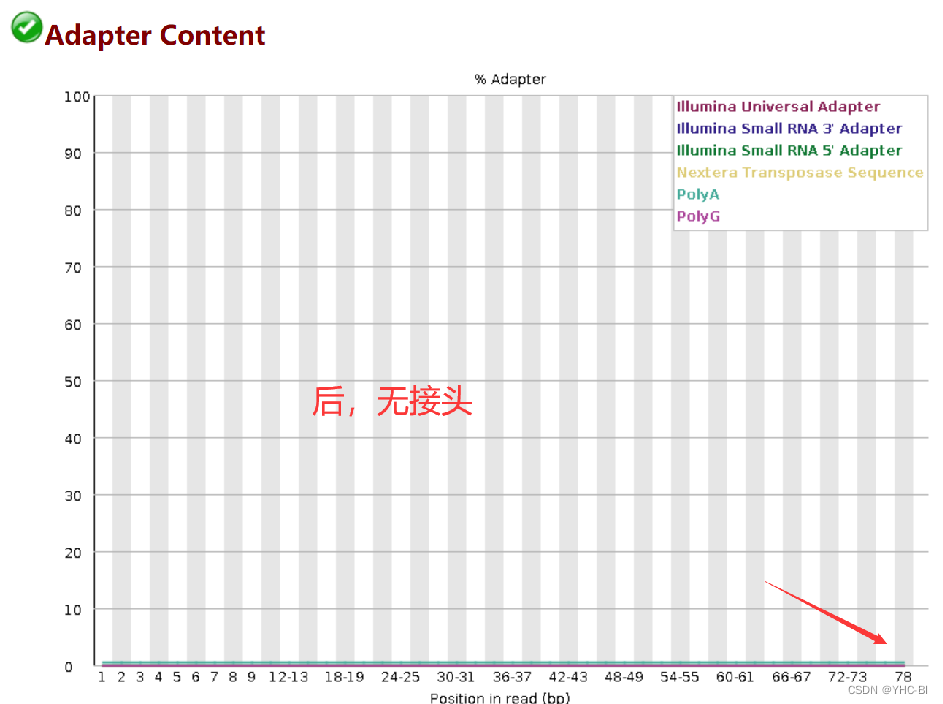
接头部分,使用肉眼就可以看出,接头别去掉了,这部分很重要,数据中的接头是要去掉的,这样不会影响后面的数据,降低噪音,提高数据质量与准确性。
其他质控部分就不看了。自己可以找个数据跑一跑。
到此,本文内容结束,这篇文章是经过了自己学习实践出来的,参考了很多资料,如若有大佬能指出错误,我将感激