基于人工智能的多肽药物分析问题(六)
2021SC@SDUSC
1. 卷积神经网络
1.1 卷积神经网络优势
在学习卷积神经网络之前,使用的是全连接神经网络,但是:
如果用全连接神经网络处理大尺寸图像具有三个明显的缺点:
(1)首先将图像展开为向量会丢失空间信息;
(2)其次参数过多效率低下,训练困难;
(3)同时大量的参数也很快会导致网络过拟合。
而使用卷积神经网络可以很好地解决上面的三个问题。
与常规神经网络不同,卷积神经网络的各层中的神经元是3维排列的:宽度、高度和深度。其中的宽度和高度是很好理解的,因为本身卷积就是一个二维模板,但是在卷积神经网络中的深度指的是激活数据体的第三个维度,而不是整个网络的深度,整个网络的深度指的是网络的层数。举个例子来理解什么是宽度,高度和深度,假如使用CIFAR-10中的图像是作为卷积神经网络的输入,该输入数据体的维度是32x32x3(宽度,高度和深度)。**我们将看到,层中的神经元将只与前一层中的一小块区域连接,而不是采取全连接方式。**对于用来分类CIFAR-10中的图像的卷积网络,其最后的输出层的维度是1x1x10,因为在卷积神经网络结构的最后部分将会把全尺寸的图像压缩为包含分类评分的一个向量,向量是在深度方向排列的。
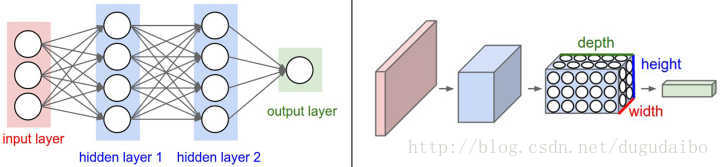
2. 卷积神经网络的各种层
卷积神经网络主要由这几类层构成:输入层、卷积层,ReLU层、池化(Pooling)层和全连接层。通过将这些层叠加起来,就可以构建一个完整的卷积神经网络。在实际应用中往往将卷积层与ReLU层共同称之为卷积层,所以卷积层经过卷积操作也是要经过激活函数的。具体说来,卷积层和全连接层(CONV/FC)对输入执行变换操作的时候,不仅会用到激活函数,还会用到很多参数,即神经元的权值w和偏差b;而ReLU层和池化层则是进行一个固定不变的函数操作。卷积层和全连接层中的参数会随着梯度下降被训练,这样卷积神经网络计算出的分类评分就能和训练集中的每个图像的标签吻合了。
更简单通俗的说法是CBAPD,卷积就是用于提取特征,起到特征提取器的作用
C 卷积层
B 批标准化
A 激活
P 池化
D 全连接层
2.1 卷积层
-
滤波器的作用或者说是卷积的作用。卷积层的参数是有一些可学习的滤波器集合构成的。每个滤波器在空间上(宽度和高度)都比较小,但是深度和输入数据一致。直观地来说,网络会让滤波器学习到当它看到某些类型的视觉特征时就激活,具体的视觉特征可能是某些方位上的边界,或者在第一层上某些颜色的斑点,甚至可以是网络更高层上的蜂巢状或者车轮状图案。
-
可以被看做是神经元的一个输出。神经元只观察输入数据中的一小部分,并且和空间上左右两边的所有神经元共享参数(因为这些数字都是使用同一个滤波器得到的结果)。
-
降低参数的数量。这个由于卷积具有“权值共享”这样的特性,可以降低参数数量,达到降低计算开销,防止由于参数过多而造成过拟合。
3.2 卷积层的大小选择
几个小滤波器卷积层的组合比一个大滤波器卷积层好。假设你一层一层地重叠了3个3x3的卷积层(层与层之间有非线性激活函数)。在这个排列下,第一个卷积层中的每个神经元都对输入数据体有一个3x3的视野。第二个卷积层上的神经元对第一个卷积层有一个3x3的视野,也就是对输入数据体有5x5的视野。同样,在第三个卷积层上的神经元对第二个卷积层有3x3的视野,也就是对输入数据体有7x7的视野。假设不采用这3个3x3的卷积层,二是使用一个单独的有7x7的感受野的卷积层,那么所有神经元的感受野也是7x7,但是就有一些缺点。首先,多个卷积层与非线性的激活层交替的结构,比单一卷积层的结构更能提取出深层的更好的特征。其次,假设所有的数据有C个通道,那么单独的7x7卷积层将会包含 C * (7*7*C)=49CC个参数,而3个3x3的卷积层的组合仅有 3(C*(3*3*C))=27C*C 个参数。直观说来,最好选择带有小滤波器的卷积层组合,而不是用一个带有大的滤波器的卷积层。前者可以表达出输入数据中更多个强力特征,使用的参数也更少。唯一的不足是,在进行反向传播时,中间的卷积层可能会导致占用更多的内存。
2.2 receptive field
中文翻译过来叫感受野
在处理图像这样的高维度输入时,让每个神经元都与前一层中的所有神经元进行全连接是不现实的。相反,我们让每个神经元只与输入数据的一个局部区域连接。该连接的空间大小叫做神经元的感受野(receptive field),它的尺寸是一个超参数(其实就是滤波器的空间尺寸)。在深度方向上,这个连接的大小总是和输入量的深度相等。需要再次强调的是,我们对待空间维度(宽和高)与深度维度是不同的:连接在空间(宽高)上是局部的,但是在深度上总是和输入数据的深度一致。
2.3 池化层
通常在连续的卷积层之间会周期性地插入一个池化层。它的作用是逐渐降低数据体的空间尺寸,这样的话就能减少网络中参数的数量,使得计算资源耗费变少,也能有效控制过拟合。汇聚层使用 MAX 操作,对输入数据体的每一个深度切片独立进行操作,改变它的空间尺寸。最常见的形式是汇聚层使用尺寸2x2的滤波器,以步长为2来对每个深度切片进行降采样,将其中75%的激活信息都丢掉。每个MAX操作是从4个数字中取最大值(也就是在深度切片中某个2x2的区域),深度保持不变。
2.4 其他
激活层一般根据不同的计算要求选择不同的激活函数,有时候可以舍弃掉批处理的部分,直接在卷积层的参数中添加激活函数,就成了
卷积-池化-卷积-池化-卷积-池化-全连接 这样的神经网络结构
2.5 层的尺寸设置规律
- 输入层 ,应该能被2整除很多次。常用数字包括32(比如CIFAR-10),64,96(比如STL-10)或224(比如ImageNet卷积神经网络),384和512。
- 卷积层 ,应该使用小尺寸滤波器(比如3x3或最多5x5),使用步长S=1。还有一点非常重要,就是对输入数据进行零填充,这样卷积层就不会改变输入数据在空间维度上的尺寸。比如,当F=3,那就使用P=1来保持输入尺寸。当F=5,P=2,一般对于任意F,当P=(F-1)/2的时候能保持输入尺寸。如果必须使用更大的滤波器尺寸(比如7x7之类),通常只用在第一个面对原始图像的卷积层上。
- 汇聚层 ,负责对输入数据的空间维度进行降采样。最常用的设置是用用2x2感受野(即F=2)的最大值汇聚,步长为2(S=2)。注意这一操作将会把输入数据中75%的激活数据丢弃(因为对宽度和高度都进行了2的降采样)。另一个不那么常用的设置是使用3x3的感受野,步长为2。最大值汇聚的感受野尺寸很少有超过3的,因为汇聚操作过于激烈,易造成数据信息丢失,这通常会导致算法性能变差。
3. 代码分析
3.1 接上周- 提取OneBodyTerms
在提取特征矩阵function中,调用了get_bond_lengths_and_angles方法计算键角和键长,氢键的三种长度和角度均在上篇博客中有所体现。
3.1.1 计算键角和键长
def get_bond_lengths_and_angles(mypose,k):
seqlen = len(mypose.sequence())
result_dict = {
}
if k > 1:
C_prev = mypose.residue(k-1).xyz("C")
N_curr = mypose.residue(k).xyz("N")
CA_curr = mypose.residue(k).xyz("CA")
C_curr = mypose.residue(k).xyz("C")
if k < seqlen:
N_next = mypose.residue(k+1).xyz("N")
if k > 1:
CpNc = N_curr - C_prev
NcCAc = CA_curr - N_curr
CAcCc = C_curr - CA_curr
if k < seqlen:
CcNn = N_next - C_curr
NcCAc_len = NcCAc.norm()
result_dict["NcCAc_len"] = NcCAc_len
CAcCc_len = CAcCc.norm()
result_dict["CAcCc_len"] = CAcCc_len
if k < seqlen:
CcNn_len = CcNn.norm()
result_dict["CcNn_len"] = CcNn_len
if k > 1:
CNCA = angle_between_vecs(CpNc.negated(), NcCAc)
result_dict["CpNcCAc"] = CNCA
NCAC = angle_between_vecs(NcCAc.negated(), CAcCc)
result_dict["NcCAcCc"] = NCAC
if k < seqlen:
CACN = angle_between_vecs(CAcCc.negated(), CcNn)
result_dict["CAcCcNn"] = CACN
return result_dict
思路分析:
初始化相关变量,首先通过计算获得相关原子"C",“N”,"CA"的xyz坐标;接着,根据蛋白质模型的长度与传入残基数目的比较,计算得到相关的原子-原子向量,接着,根据计算所得到的原子-原子向量的的坐标,计算分布在其中的键的长度,计算完毕后,返回map,其中共有6个键值对保存着蛋白质的特征
之后,对于第一个和最后一个残基,将feature_dict的相应键赋值为0。并返回处理后的数据。
3.1.2 能量关系
调用了pyrosetta的一个能量函数get_fa_scorefxn(),用于计算接下来的能量矩阵,将pose,得分函数和一组分数项常量传入方法,
分数项分为"p_aa_pp", “rama_prepro”, “omega”, “fa_dun”,查阅了一下相关的资料,这四类的介绍分别如下:
其中:
p_aa_pp: 通过贝叶斯定理进行的转化统计所得项,含义是在目前的phi/psi区间内,出现某一种氨基酸的概率。用于评估氨基酸的可替换性,与突变设计相关的能量项,也可以理解为设计得到的氨基酸与骨架匹配程度的一个评估。
rama_prepro:直接从数据库中得到的统计量,其含义为给定一种氨基酸类型时,其骨架二面角的概率分布,分为i+1为脯氨酸和非脯氨酸两种情况。如果模型中二面角落于频率分布高的格点区域,则能量越低,骨架构象合理的可能性越高。当偏离这些高频率分布的区域时,骨架二面角的能量越高。
omega:肽键二面角会根据phi/psi角度变化,但基本固定在180°(trans-)或0°(cis-),当肽键二面角偏离时,生成惩罚。
fa_dun:氨基酸残基内部的原子的冲突。
能量函数说明:
在Rosetta中评估一个模型的好坏,最直观的方法就是使用Rosetta的打分系统进行评估,也就是常说的能量函数。顾名思义,我们通过一些与能量直接相关的打分项对蛋白质的结构坐标进行打分的过程。Rosetta能量函数由一系列可衡量的几何统计或经典物理相互作用能量经过加权后得到的函数形式。在给定原子坐标的条件下,评估原子之间的相互作用能量的大小。
def get_one_body_score_terms(pose, scorefxn, score_terms):
one_body_score_terms = []
scorefxn(pose)
energy_obj = pose.energies()
for pos in range(1, len(pose.sequence()) + 1):
energy_string = get_energy_string_quick(energy_obj, pos)
energy_dict = energy_string_to_dict(energy_string)
res_scores = []
for term in score_terms:
res_scores.append(energy_dict[term])
one_body_score_terms.append(res_scores)
return np.array(one_body_score_terms).T
对蛋白质序列作循环,对每个蛋白质序列元素调用get_energy_string_quick方法,获取以字符串形式存储的能量名称,通过传入energy对象和残基的索引号,可以快速获取,之后再根据获取到的string 调用energy_string_to_dict方法,将字符串映射为浮点数,便于之后数据放入模型中进行训练。
def energy_string_to_dict(energy_string):
# given an energy_string
# returns a dictionary (string --> float) of ALL energy terms
energy_string = energy_string.replace(") (", ")\n(")
energy_string = energy_string.replace("( ", "").replace(")", "")
energy_list = energy_string.split("\n")
energy_dict = {
}
for element in energy_list:
(score_term, val) = element.split("; ")
energy_dict[score_term] = float(val)
return energy_dict
3.1.3 定义输出
prop = extract_AAs_properties_ver1(aas)
np.savez_compressed(outfile,
idx = pdict['idx'],
val = pdict['val'],
phi = pdict['phi'],
psi = pdict['psi'],
omega6d = pdict['omega6d'],
theta6d = pdict['theta6d'],
phi6d = pdict['phi6d'],
tbt = _2df,
obt = _1df,
prop = prop,
maps = maps)
if verbose: print("Processed "+filename+" (%0.2f seconds)" % (time.time() - start_time))
except Exception as inst:
print("While processing", outfile+":", inst)
这里将之前所有的数据全部存储到outflie文件中,并给出debug语句。
3.2 多处理器情况
if num_process == 1:
for a in arguments:
pyErrorPred.process(a)
else:
pool = multiprocessing.Pool(num_process)
out = pool.map(pyErrorPred.process, arguments)
这边利用了python的进程池,调用了multiprocessing.Pool。当被操作对象数目不大时,可以直接利用multiprocessing中的Process动态成生多个进程,十几个还好,但如果是上百个,上千个目标,手动的去限制进程数量却又太过繁琐,此时可以发挥进程池的功效。
Pool可以提供指定数量的进程供用户调用,当有新的请求提交到pool中时,如果池还没有满,那么就会创建一个新的进程用来执行该请求;但如果池中的进程数已经达到规定最大值,那么该请求就会等待,直到池中有进程结束,才会创建新的进程来它。
使用了out = pool.map(pyErrorPred.process, arguments)加快处理过程
3.3 进行预测
之前的博客加上以上部分讲述了模型训练的思路及过程,接下来是预测过程
def predict(samples, distogram, modelpath, outfolder, verbose=False, transpose=False):
n_models = 2
for i in range(1, n_models):
modelname = modelpath+"_rep"+str(i)
if verbose: print("Loading", modelname)
model = Model(obt_size=70,
tbt_size=58,
prot_size=None,
num_chunks=5,
optimizer="adam",
mask_weight=0.33,
lddt_weight=10.0,
name=modelname,
verbose=False)
model.load()
for j in range(len(samples)):
if verbose: print("Predicting for", samples[j], "(network rep"+str(i)+")")
tmp = join(outfolder, samples[j]+".features.npz")
batch = getData(tmp, outfolder, distogram)
lddt, estogram, mask = model.predict2(batch)
if transpose:
estogram = (estogram + np.transpose(estogram, [1,0,2]))/2
mask = (mask + mask.T)/2
np.savez_compressed(join(outfolder, samples[j]+".npz"),
lddt = lddt,
estogram = estogram,
mask = mask)
首先调用模型,实例化model对象,这里的model使用的是tensorflow框架,调用load方法加载模型,在load方法中,将tensorflow训练中所用到的所有参数全部保存到model.ckpt文件。
之后调用getData()方法将之前提取出的一维,二维,三维features全部加载到内存。
对于给定的一个batch的数据,调用了predict2方法
def predict2(self, batch):
f3d, f1d, f2d = batch
if not self.feature_mask is None:
f1d[:, self.feature_mask[0]] = 0
f2d[:, :, self.feature_mask[1]] = 0
if self.ignore3dconv:
feed_dict = {
self.ops["obt"]: f1d,\
self.ops["tbt"]: f2d}
else:
feed_dict = {
self.ops["obt"]: f1d,\
self.ops["tbt"]: f2d,\
self.ops["idx"]: f3d[0],\
self.ops["val"]: f3d[1]}
operations = [self.ops["lddt_predicted"],
self.ops["estogram_predicted"],
self.ops["mask_predicted"]]
return self.sesh.run(operations, feed_dict=feed_dict)
根据使用者的需要使用或者忽略三维特征,使用该模型。最后将预测数据输出到相应的文件中。