1. 回顾超几何分布的定义
一般地,假设共有N件产品,其中M件次品,现在从中抽取n件做检查,抽到k件次品的概率分布服从超几何分布。
数学表达式如下:
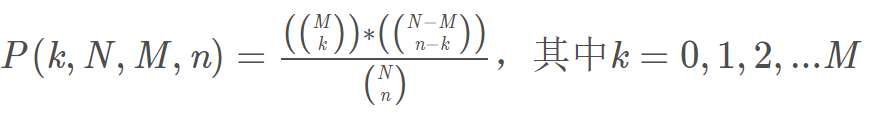
2. 超几何分布的一个应用:求解overlap显著性
- 以一个实际的生物实验为例。
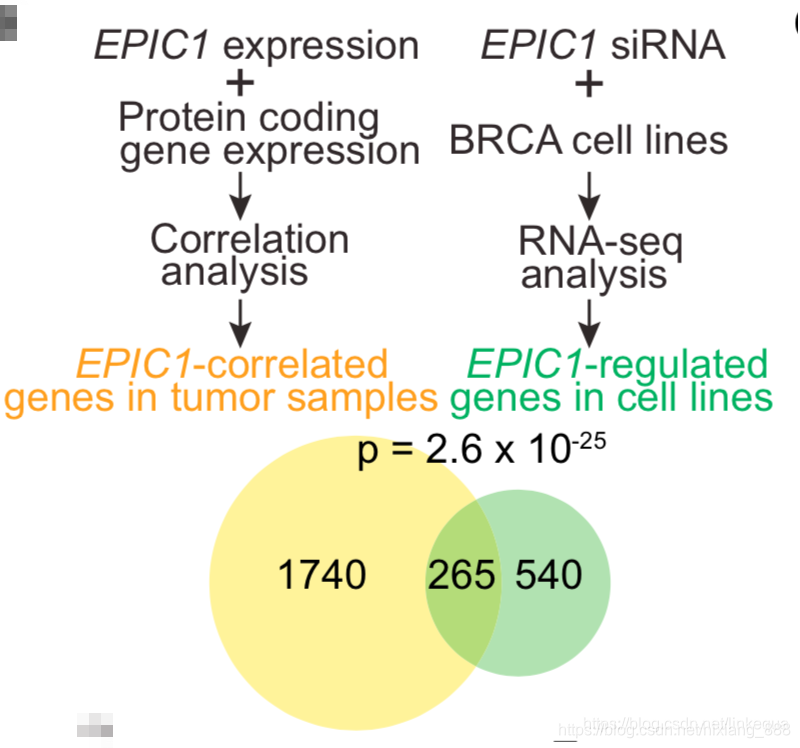
在上述实验中,我们假设总共的基因个数为20000个(假定为产品的总数),图中左边圈的总数为2005(1740+265)个(假定为次品的总个数),现从中抽取805(540+265)个,需要计算得到基因的个数(次品数量)大于等于265的概率。
思考过程:在次品个数是少数的情况下,overlap越高,从超几何分布来看,发生的概率越小。现在的overlap是265,可能会是过高的那种情况,那么现在计算overlap是265以及大于265的概率之和,如果这个概率很小,那就说明发生265这个事件不是随机的,进而就推出来了overlap为265是显著性高的一个事件。
# 用R语言计算为:
phyper(265-1, 1740+265, 20000-(1740+265), 265+540, lower.tail=F)
# Note: lower.tail使用的逻辑为:
# if TRUE (default), probabilities are P[X ≤ x], otherwise, P[X > x].
3. GO/KEGG的富集分析
假设背景基因有20000个基因,分布于若干个GO/KEGG-term中,其中,关注的某一个A-term中,含有342个基因。现我们获得了244个显著差异表达的基因,其中有17个基因富集于A-term中,请问我们获得的差异基因集是否显著富集于A-term中?
- 分析
| - | 获得差异基因数目 | 差异基因集中富集于A-term中的基因数量 | 背景基因数量 | 背景基因集中富集于A-term的基因数量 |
|---|---|---|---|---|
| A-term | 244 | 17 | 20000 | 342 |
| hypergenomic parameter |
k | x | m+n | m |
注解:
- x: 在不放回抽样中,所抽取的基因集中,富集于A-term中的基因数量
- m: 在背景基因集中,A-term中的基因数量
- n: 在背景基因集中,非A-term中的基因数量
- k: 在放回抽样中,所抽取的基因中基因的数量
- 计算
from scipy import stats
#需要注意的是16是由17-1得到的
stats.hypergeom.sf(16,20000,342, 244)
# --------------------------------------------------- #
# R中的实现方式
phyper(x, m, n, k, lower.tail=FALSE)