小罗碎碎念
这篇文章是一篇综述,标题为《Profiling cell identity and tissue architecture with single-cell and spatial transcriptomics》,发表在《Nature Reviews Molecular Cell Biology》上。

文章讨论了单细胞转录组学(single-cell transcriptomics)和空间转录组学(spatial transcriptomics)技术的最新进展、挑战和前景,特别关注了这些技术在识别和表征细胞状态以及多细胞邻域方面的应用。
文章的主要内容包括:
-
单细胞转录组学:讨论了单细胞RNA测序(scRNA-seq)如何改变了我们对细胞多样性和基因表达动态的理解,并讨论了样本处理、数据整合、识别微妙的细胞状态、轨迹建模、去卷积和空间分析等方面的进展。
-
空间转录组学:探讨了空间转录组学如何将单细胞置于多细胞邻域中,并识别空间上重现的表型,即生态型(ecotypes)。
-
临床转化:讨论了单细胞和空间转录组学在临床研究中的应用,包括干细胞生物学、免疫学和肿瘤生物学。
-
人工智能和单细胞转录组学的未来:讨论了深度学习等人工智能技术在分析单细胞和空间转录组学数据中的应用。
-
结论:总结了单细胞转录组学和空间转录组学技术如何革新我们对细胞状态在复杂组织中起源、调节和维持的理解。
文章强调了这些技术在生物医学研究中的重要性,并展望了它们在临床应用中的潜力。同时,文章也指出了在实验设计和生物信息学分析中存在的挑战,并提出了一些可能的解决方案。
一、引言
自单细胞RNA测序(scRNA-seq)技术问世近15年以来,该技术已深刻改变了我们对细胞异质性和基因表达动态的理解,涵盖了多种物种、组织和生命阶段。此方法促进了大型细胞图谱的构建、罕见细胞类型和状态的发现,以及复杂生物过程(如早期胚胎发育、大脑发育和癌症生长)中细胞轨迹的绘制1。
单细胞水平的RNA分析达到了前所未有的规模和分辨率,可与基因组学和蛋白质组学分析相结合,并在原位测量,以揭示组织内相邻细胞之间的复杂空间组织和功能关系2,3。
人类细胞图谱作为收集跨组织和物种的单细胞RNA测序观测数据任务的一部分,常被比作开创性的人类基因组计划,这一类比强调了其在推动技术发展、揭示生物多样性以及改善疾病治疗方面的潜力4,5。
通过本篇推文,我们可以一起了解单细胞转录组学在样本制备、数据整合与校正、大型细胞图谱的编纂、罕见和细微细胞状态识别、推断细胞轨迹以及预测发育潜能方面的最新进展和挑战(图1和表1)。随后,我们将探讨空间转录组学(ST)的技术和生物学考量,以及将scRNA-seq与ST整合的方法(表1)。

我们引入了空间生态型的概念,即空间上共定位且表型一致的一组细胞状态,并讨论了其在表征正常和病变组织微环境方面的相关性(图1)。

这张图展示了从实验设计到生物学和临床发现的整个研究流程,特别是单细胞转录组学和空间转录组学的应用。
- 实验设计(Experimental design):研究从不同器官和物种中编目单细胞和空间转录组数据
- 单细胞转录组学(Single-cell transcriptomics):
- 细胞(Cells):从组织中分离出单个细胞。
- 单细胞RNA(Single-cell RNA):提取每个细胞的RNA。
- 细胞类型和状态(Cell types and states):分析RNA以确定细胞的类型和状态。
- 细胞轨迹(Cellular trajectories):通过分析细胞状态的变化来推断细胞的发育轨迹。
- 空间转录组学(Spatial transcriptomics):
- 多细胞邻域(Multicellular neighbourhoods):研究细胞在组织中的分布和相互作用。
- 空间生态型(Spatial ecotype):定义细胞在空间上的分布模式。
- 机器学习和人工智能(Machine learning and artificial intelligence):利用这些技术来分析和解释大量的数据,从而揭示细胞间的信号传递和相互作用。
- 生物学和临床发现(Biological and clinical discoveries):最终,这些研究有助于新的生物学理解和临床应用的开发。
整体而言,这张图强调了单细胞和空间转录组学技术在解析细胞多样性、组织结构和功能方面的重要性,以及这些技术如何通过机器学习和人工智能的应用来促进生物学和医学的进步。
随着scRNA-seq和ST产生大量高维数据,分解方法和更简单的检测手段,如免疫组化和基因表达特征,可以促进其向大型临床队列的转化。
此外,随着单细胞基因组数据规模和复杂性的指数级增长,我们还会在推文的末尾一起探讨基础模型在单细胞生物学中的一些潜在应用,并分析如何将人工智能技术与单细胞和空间转录组学结合起来。
二、单细胞转录组学
尽管单细胞RNA测序(scRNA-seq)技术已彻底改变了我们对多细胞生物学的理解,但对样本制备、数据规范化和批次整合影响的忽视可能导致错误的结论和对结果的误解。
2-1:在样本处理和细胞解离中识别技术和生物学协变量
样本制备、细胞解离和RNA捕获是生成scRNA-seq数据的第一步,也是标准化程度最低的步骤(图2a)。

这张图片展示了样本处理的流程,特别是针对单细胞测序(single-cell sequencing)的样本准备步骤。流程如下:
- 组织采集:从生物体(如人体)获取组织样本。
- 死后时间间隔(Post-mortem interval):指组织从生物体取出到开始处理之间的时间,这个时间间隔可能会影响样本的质量。
- 组织样本:取出的组织样本,准备进行后续处理。
- 福尔马林固定石蜡包埋(FFPE):一种组织保存方法,通过固定和包埋入石蜡来保存组织样本,便于切片和长期保存。
- 组织特异性化学品:使用特定化学品处理组织,以帮助分离细胞或提取RNA。
- 机械消化(Mechanical digestion):通过物理方法破坏组织结构,分离出单个细胞。(例如研磨)
- 化学消化(Chemical digestion):使用化学方法进一步分解组织,可能包括酶解等步骤来提取RNA。
- 单细胞测序(Single-cell sequencing):对单个细胞进行测序,分析其基因表达和其他分子特征。
这个流程是单细胞转录组学研究中的关键步骤,确保从组织样本中获取高质量的单细胞RNA,为后续的生物信息学分析提供基础。
生物体死亡后,转录和其他细胞变化立即发生。在数分钟至数小时内,由于免疫细胞渗出机制的衰退和脆弱细胞的凋亡,组织成分发生变化6,7。RNA合成停止,减少了每个细胞可检测到的转录本数量8,并发生非随机的转录改变,包括线粒体RNA含量、剪接失调和替代异构体增加的组织特异性变异,以及与碳水化合物代谢、免疫反应、细胞周期、应激反应和细胞坏死的基因失活6,7,9。使用不同的解离协议时,也会观察到类似的时间依赖性变化,这取决于消化酶的类型和解离的孵化温度10。
在解离过程中,组织的内在特性和细胞的内在特性也可能影响细胞组成。组织中的化学物质,如胃酸、胆酸和肠胰酶,可以在分离过程中直接裂解细胞并降解RNA11–13。难以分离的粘附细胞,如成纤维细胞14,具有丝状伪足的大细胞,如巨噬细胞和脂肪细胞15,以及寿命短的细胞,如中性粒细胞16,在解离过程中可能被轻易错过或过滤掉。
酶消化也会降解细胞表面抗原,从而在流式细胞术及其他相关方法(如CITE-seq和REAP-seq17–20)中降低抗体介导的检测性。这些细胞和转录变化可能对下游分析产生深远影响,如低维嵌入、聚类、差异基因表达和谱系追踪8。
为减轻这些解离偏差,已开发出实验策略,但它们又引入了新的混淆因素。
单核RNA测序(snRNA-seq)从冷冻和难以解离的组织(如神经元和胶质细胞)中分离核RNA。然而,从不同的生物体、组织和感兴趣的细胞中分离核仍然需要定制化的协议来最小化解离偏差21。
转录分析用的核分离易受到环境RNA的污染22,并排除了关键的细胞质转录本,包括参与小胶质细胞活化23、存储在处理体中的调节性RNA24,以及细胞在有核膜缺失的分裂期间核RNA14。
化学固定可以立即保存感兴趣组织的细胞和转录内容,但它也破坏了细胞及其内容的超微结构,使得全转录组测序变得困难。10x Genomics Flex Kit(https://go.nature.com/4crkIB3)使用预定的探针组从小鼠和人类固定组织中提取RNA。尽管此协议最小化了样本处理和技术批次对数据质量的影响,但RNA定量仅限于精选的基因组区域,单细胞核的解离仍然容易受到细胞组成扭曲的影响。
下面将详细讨论的空间转录组学(ST)可以在不解离的情况下保存细胞组织背景,可能提供更生理代表性的结果。
减少样本制备时间、优化消化缓冲液和为这些预处理步骤制定社区标准,对于最小化实验变异性至关重要。使用计算工具对技术协变量进行事后校正可以提高scRNA-seq数据的生物学相关性,但这并不能消除详细记录预处理变量和设计对照组以准确消除混淆因素而不减弱生物学信号的需求。
2-2:计算策略用于样本整合和批次校正
在涉及多个样本的单细胞实验中,样本间的感兴趣生物学差异可能被无意的技术和生物学协变量所掩盖,这些协变量会影响下游分析。
如前所述,样本制备和处理的变化,以及与测序相关的因素,如文库大小、PCR循环次数和测序仪器,都可能在数据中引入人工产物,导致单细胞更多地按技术批次而非生物学相似性聚类25。
与受试者相关的其他因素,如样本个体的年龄、性别和血统26,或特征异质性,如细胞周期阶段27或线粒体RNA含量28,也可能掩盖感兴趣的生物学变量。为纠正批次特异变异同时保留真实生物学信号,已开发出许多计算方法(见表1)。
ComBat30和limma31是用于微阵列和批量RNA-seq数据的流行线性调整模型,但由于单细胞RNA测序(scRNA-seq)数据的稀疏性和批次间细胞组成的变异性,它们在scRNA-seq数据中的应用具有挑战性。这一限制促使了如相互最近邻(MNN)等方法的发展,该方法跨批次识别相似细胞的邻域,并使用这些对齐来整合批次并校正基因表达数据32(见图2b)。
这张图展示了数据整合和批次校正(batch correction)的过程,特别是在单细胞转录组学数据分析中。

这张图分为两个部分,展示了单细胞转录组学数据的整合和批次校正的过程。
上半部分:数据整合
- 左侧:显示了两个不同批次(Batch 1 和 Batch 2)的单细胞数据在二维空间(Dimension 1 和 Dimension 2)中的分布。每个批次的细胞群体用不同颜色表示,显示出明显的批次间差异。
- 右侧:展示了经过数据整合后的结果,两个批次的数据点更加混合,减少了批次间的技术变异,使得相似的细胞群体聚集在一起。
下半部分:批次校正
- 左侧:展示了基因表达矩阵,其中列代表基因,行代表细胞。颜色变化表示基因表达水平,不同批次的表达模式存在差异。
- 右侧:经过批次校正后,基因表达矩阵中的颜色分布更加均匀,表明批次效应被校正,使得数据更加准确地反映生物学差异而非技术噪声。
整体而言,这张图强调了在单细胞转录组学数据分析中进行数据整合和批次校正的重要性,这有助于消除或减少由于实验设计或技术因素导致的批次效应,从而更准确地揭示细胞间的生物学差异。
为加速跨批次寻找共享元素的过程并提高对噪声的鲁棒性,可以使用主成分分析(例如,fastMNN32、BBKNN33、Harmony34)、奇异值分解(例如,Scanorama35)、整合非负矩阵分解(例如,LIGER36)或典范相关分析(例如,Seurat37)等方法将基因表达轮廓降至低维嵌入,以更有效地识别和对齐相似细胞。
这些方法可能需要用户输入和参数调整,因此鼓励使用先验知识和不过分依赖默认设置的试错策略以获得最佳结果。尽管数据整合工具改善了可视化、聚类和轨迹构建,但高效准确地校正原始高维基因表达数据矩阵仍然具有挑战性。
**分析批次校正后的基因表达水平通常并不可取,因为这可能消除真正的生物学差异(称为“II型错误”),同时在数据中引入虚假的差异表达基因(称为“I型错误”)**38。
为跨多个样本进行比较,已提出替代策略。
伪批量分析将单个细胞的基因表达聚合成群组级值,将每个样本视为重复39。这最小化了单细胞间的稀疏性、噪声和共线性,但忽略了细胞簇内的异质性、细胞丰度和批次间变异性。协变量和混合效应建模是估计不同因素对单细胞基因表达数据影响的其他方法38。
非参数方法,如Wilcoxon秩和检验,不假设底层分布,但通常限于简单实验设计和两组比较40。在批次同时包含病例和对照样本或在不同样本内而非跨样本进行差异基因表达分析的情况下,跨组进行基因级统计的元分析是一种合理的策略37,39。还出现了无需预定义细胞亚群的无聚类策略,它们可以跨批次识别差异丰富的细胞状态41。
基于神经网络的技术42–46已显示出在低维嵌入空间和原始基因表达空间中对大量批次进行对齐的可扩展性和速度提升。这些多任务技术可以在整合和校正的空间中有效地执行数据规范化、聚类、降维和差异基因表达分析。如最近一篇预印刊报导,批次校正方法存在过度校正和消除所需生物学信号的风险47。
一种保留生物学信号的策略是使用半监督方法,这些方法可以由细胞注释信息指导。半监督深度学习方法,包括scANVI和scGEN,在批次校正和保留生物学变异方面表现优于无监督方法48。
仅凭计算工具可能不足以完全区分生物学信号和混淆变量。需要对scRNA-seq实验的设计进行标准化,并主动包含技术对照组,从中可以学习批次效应并应用于案例(例如,“控制池”49),以准确建模跨样本变异49,50。
2-3:构建和整理大型单细胞图谱
近年来,由于多个涉及多种组织和生物的大型单细胞图谱项目的开展,我们见证了转录谱单细胞数量的指数级增长。值得注意的是,约80%的超过1亿2800万个单细胞转录组是在过去3年内测序的,涵盖了至少190种组织和76个物种51。
这些图谱创建项目的最终目标是构建一个注释完备、公开可访问的细胞类型和状态词典,该词典可作为细胞标记、转录特征、疾病关联和假设生成的参考52,53(见图2c)。
这张图展示了细胞图谱(cell atlases)的构建过程。细胞图谱是一种资源,它详细描述了不同细胞类型在生物体内的分布和特性。
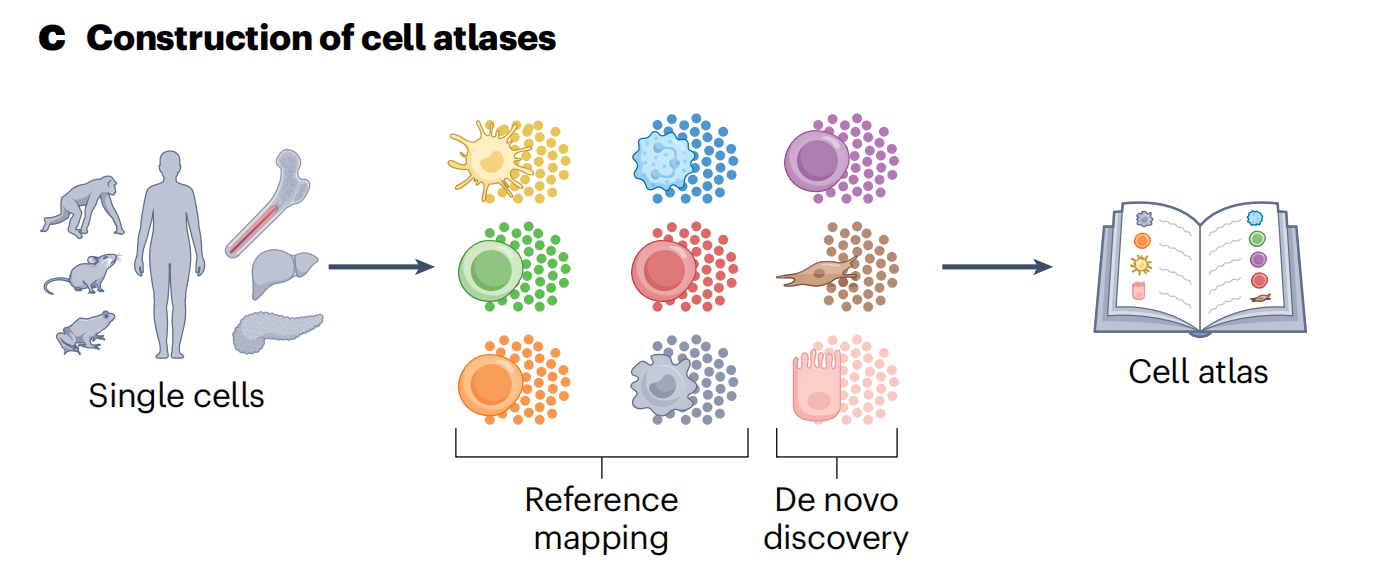
构建过程包括以下几个步骤:
-
单细胞(Single cells):从不同的生物体或组织中获取单细胞样本。
-
参考映射(Reference mapping):将单细胞数据与已知的细胞类型进行比较,以识别和分类细胞。
-
新发现(De novo discovery):除了参考映射外,还会发现新的、未知的细胞类型或状态。
-
细胞图谱(Cell atlas):将所有这些信息汇编成一本“图谱”,提供对细胞多样性和组织分布的全面视图。
整体而言,这张图强调了通过单细胞分析来构建细胞图谱的重要性,这有助于我们更好地理解生物体内细胞的复杂性和组织结构。
尽管近期取得了进展,但质量控制和数据整合仍然面临着样本处理和测序方式可变性的挑战48。为解决这些问题,正在通过人类肿瘤图谱网络(Human Tumor Atlas Network)等大规模努力,对单细胞实验和计算工作流程进行规范化,特别是肿瘤样本54。
注释单细胞数据存在多种策略,包括使用参考数据的标记基因检测55–57、基因集富集分析58、基于相关性的技术59–62、通用分类器63–66和最大似然估计器67等(见表1)。
大型语言模型,如GPT4,可以基于用户提供的标记基因等提示链轻松注释细胞68。此外,正在开发使用深度学习模型的迁移学习策略,以将单细胞参考中的注释映射到小型scRNA-seq数据集,并发现疾病特异性细胞状态,例如COVID-19和特发性肺纤维化患者70,71。
单细胞参考整理的挑战包括需要更详细的细胞级和样本级注释,建立涵盖各种组织、器官和疾病状态的统一细胞状态本体论72,以及精确地将查询数据集与条件匹配的参考图谱对齐。
在人类参考图谱中,必须特别注意确保包含广泛的 demograhic因素,如年龄、性别/性别、种族和民族73,以及多样的生理状态和病理条件。
为促进查询数据集中新细胞类型的发现,还需谨慎避免将细胞不适当地对齐到预先存在的细胞标签。一种可能的选项是,如果无法将细胞确定性地分配到任何已建立的参考细胞类型,则将其分配到“新颖”类别74。
2-4:实验和计算策略在scRNA-seq中识别稀有和微妙细胞状态
scRNA-seq的主要目标是识别在批量样本中无法检测到的稀有和微妙细胞群体。
尽管scRNA-seq已导致组织驻留干细胞和祖细胞75、疾病驱动免疫细胞76和肿瘤细胞谱系状态77,78的发现,但当前的实验和计算方法在识别稀有和微妙细胞状态方面仍面临许多挑战。
如上一节所述,严苛的解离条件可能消除脆弱细胞或诱导与细胞状态相关的应激反应,导致在预处理步骤中丢失稀有和微妙细胞群体14,79,80(见图2d)。
这张图展示了识别稀有和微妙细胞状态的流程,特别是在单细胞研究中的应用。

流程包括以下几个步骤:
-
样本处理(Sample processing):
- 从组织中分离出多种细胞类型。
- 经过解离散(Dissociation)步骤,导致细胞组成和基因表达的变化。
-
基因选择(Gene selection):
- 通过表达矩阵选择与稀有或微妙细胞状态相关的基因。
- 例如,Hoxb5基因可能被选为标记特定细胞状态的基因。
-
平台选择(Platform selection):
- 根据所需的细胞数量、基因数量以及是否需要全长测序,选择合适的单细胞测序平台。
- 例如,Droplet平台适合高细胞数、低基因数、非全长测序;而Targeted平台适合低细胞数、高基因数、全长测序。
-
细胞富集(Enrichment):
- 解剖富集(Anatomic enrichment):使用激光显微镜选择特定位置的细胞。
- 细胞计量富集(Cytometric enrichment):通过流式细胞仪物理分离特定细胞。
整体而言,这张图强调了在单细胞转录组学研究中,如何通过样本处理、基因选择、平台选择和细胞富集等步骤来识别和研究稀有和微妙的细胞状态。这些步骤对于揭示细胞的复杂性和异质性至关重要,有助于深入理解组织功能和疾病机制。
例如,标准解离协议诱导独特的应激反应并激活静止肌肉干细胞,导致静止干细胞群体表征不佳,并将这种解离人工产物误分类为新细胞状态79,81。
稀有和微妙细胞状态识别的另一个主要因素是它们在整个研究群体中的频率,以及测序细胞数量是否足以检测它们。例如,组织驻留干细胞极为罕见,即使是未经纯化的组织的 large-scale scRNA-seq 也可能在没有靶向富集策略的情况下遗漏这些细胞82–84。
例如,Hoxb5基因的表达标记了具有高再生能力的小鼠长期造血干细胞(LT-HSCs)的一个亚群85(见图2d)。Hoxb5+ LT-HSCs在骨髓中的估计频率为0.001%,因此,在无需事先富集的条件下,要从小鼠骨髓中分离出10个 Hoxb5+ LT-HSCs,大约需要一百万个有核细胞86。
通过针对特定细胞表面标记物(如KIT和SCA-1)进行富集,同时去除表达成熟细胞表面标记物的细胞,Hoxb5+ LT-HSCs的频率增加了1000倍,显著提高了其在scRNA-seq数据中的可检测性83,87。其他scRNA-seq研究也利用了已知的细胞分布生物特征,如表面标记物、解剖位置和胚胎阶段,来分离并功能验证人类神经88和骨骼89干细胞(见图2d)。
识别稀有和微妙细胞状态通常取决于检测细胞周期、分化轨迹或代谢状态中的适度但可重复的差异。
为此,基于板的全长测序虽然比基于液滴的短读测序成本更高、耗时更长,但它提供了从单个细胞更深更广的RNA捕获90(见图2d)。
此外,全长序列提供的较低dropout率和准确的读数估计保留了对细胞间微小但关键的转录差异的解析。最近基于板技术的进展,如MAS-seq和VASA-seq,已经扩展了转录组覆盖范围,包括非编码和选择性剪接的转录异构体,这揭示了在肿瘤浸润性T细胞分化和小鼠发育过程中罕见转录本的异质分布91,92。
在捕获候选细胞及其转录本后,单细胞转录组被解析以寻找指示生物学异质性的关键特征(见表1)。正确的特征选择对于识别稀有或微妙细胞状态至关重要,因为在这一步骤中遗漏关键标记基因可能会损害细胞状态的发现。
许多分析流程使用分散性,即数据集中单个细胞间基因表达水平的变化,来优先考虑基因93–95。然而,这些测量可能会受到无关协变量的影响,并且可能对来自稀有或微妙不同细胞状态的生物学信号不敏感。在筛选出高分散性的基因后,各种计算工具应用层次聚类(例如,RaceID393)、密度测量(例如,FiRE94)或簇间距离(例如,GapClust95)来寻找稀有或微妙细胞状态。
其他特征选择策略已被使用,并与各种下游聚类策略结合以识别稀有或微妙细胞状态。
例如,GiniClust396基于基尼系数选择稀有细胞基因,该系数衡量细胞间基因表达的不平等。CellSIUS97通过生成在预定义簇中高度相关且差异表达的基因集来进行特征选择。scPNMF98使用投影非负矩阵分解(NMF)来揭示细胞和基因表达模式,也使用相关性筛选和多模态分布来选择生物学相关的基因。
需要注意的是,当使用相同的数据进行聚类后再进行差异表达分析时,细胞状态标记物假发现的风险会增加。因此,已经开发了统计方法,与上述工具结合使用,以识别更多生物学相关的标记基因99,100。
深度生成模型,如scLDS2,也正在开发中,它们可以灵活地学习数据集中单细胞的分布,并使用可解释的分类器来推断稀有细胞。然而,数据集在大小、批次和条件上的复杂性和多样性,在跨样本数据整合和校正后识别稀有细胞状态时构成了重大挑战。SEAcells101使用自适应高斯核来捕捉主要的变化源,然后在降维图上应用原型分析来识别生物学上连贯的细胞群,或元细胞。
随着单细胞数据集中注释的细胞状态越来越多,通过在单细胞RNA测序实验中展示它们的一致性,并在实验室环境中功能复现,来验证这些细胞状态定义变得至关重要。如下一节所述,空间测序为在原生微环境中情境化新细胞状态提供了新的机会,并利用空间特征来改进细胞分类学102。
2-5:追踪单个细胞在生物学转化过程中的变化
单细胞RNA测序(scRNA-seq)分析已能够精确描绘细胞在发育、分化、细胞周期以及对基因组学和代谢学扰动响应等动态生物学过程中的转录路径。
使用转录数据预测研究的单个细胞在过渡细胞状态连续体中的相对顺序是一项重大的生物学挑战。为预测不同生物学过程中的单个细胞顺序,已开发出多种计算和基因组方法(见表1)。其中一部分方法专注于预测单细胞生成其他细胞的可塑性,即从单细胞转录组学数据中预测细胞潜能。
1、轨迹推断工具
已开发出多种计算方法来推断细胞轨迹(见图3a),这张图展示了细胞轨迹的概念,这是单细胞转录组学分析中用来描述细胞状态转变和发育路径的方法。

图中的散点代表不同的细胞,它们在两个维度(Dimension 1 和 Dimension 2)上的分布显示了细胞状态的变化。
-
轨迹线(Cell trajectories):图中的曲线表示了细胞状态的转变路径。这些路径揭示了细胞如何从一个状态过渡到另一个状态,反映了细胞的发育轨迹或响应。
-
循环轨迹(Loop):图中有一个明显的循环轨迹,这可能表示某些细胞状态的循环过程,如细胞周期中的不同阶段或细胞在特定微环境中的可逆变化。
-
维度(Dimension 1 和 Dimension 2):这两个维度是通过降维技术(如PCA或t-SNE)从高维基因表达数据中提取的,用于可视化和分析细胞状态的变化。
整体而言,这张图通过细胞轨迹的可视化,帮助我们理解细胞在发育过程中的状态变化和动态行为,这对于研究细胞分化、重编程和疾病发展等生物学问题至关重要。
除了一些例外,这些轨迹推断工具涉及生成低维嵌入并通过基于图形或聚类的方法识别的节点来描绘路径。轨迹推断工具具有模块性,主要在维度减少、聚类和轨迹建模的方法上有所不同103。对于这些方法的广泛基准测试和全面讨论,参见参考文献104。
随着数据集在大小和复杂性上的增长,出现了几个挑战。
轨迹推断工具容易受到非预期协变量对维度减少和聚类的影响,并且它们依赖于准确的数据整合和批次校正来绘制跨多个样本的轨迹。
在大型、异质性的数据集中,多个细胞谱系可能同时参与不同的动态过程(例如,细胞周期和分化)。轨迹推断工具预期能够将这些复杂数据集简化为生物学部分,并在不同谱系的无关细胞或静态状态的细胞之间不强制连接的情况下绘制轨迹。
这些方法还需要用户进行大量的操作和参数调整,以识别最佳轨迹并确定动态过程的根或终端状态。
已开发出几种方法来无偏地确定转录进化的方向(见图3b)。
这张图展示了RNA和蛋白质速度的概念,这是单细胞转录组学中用来分析细胞状态转变和发育轨迹的方法。

图的左侧:
- 描述了mRNA剪接(spliced mRNA)和未剪接(non-spliced mRNA)的丰度变化,以及它们如何转化为蛋白质(protein)。
- 未剪接的mRNA首先被剪接,然后转化为蛋白质。
图的右侧:
- 展示了细胞状态的转变过程,其中每个小圆圈代表一个细胞状态,箭头表示细胞状态的变化方向。
- 颜色编码表示不同类型的mRNA和蛋白质的丰度:灰色代表未剪接的mRNA,红色代表剪接的mRNA,灰色代表蛋白质。
- 细胞状态的变化可能受到多种因素的影响,包括基因表达的变化、细胞周期的不同阶段或对外部信号的响应。
整体而言,这张图强调了通过测量mRNA和蛋白质的动态变化来推断细胞的发育轨迹和状态转变,这对于理解细胞如何响应生物学过程和疾病状态至关重要。
RNA速度分析通过比较新转录的前mRNA与成熟mRNA来估计细胞转换的速度和方向105,106;protaccel还结合了蛋白质速度,当有multi-omics数据时可提供更丰富的细胞状态动态洞察107。
类似的分子动力学也可以通过使用NASC-seq108和scSlam-seq109对新生RNA进行代谢物标记直接测量。这些工具捕获的是短时间尺度(几分钟到几小时)内发生的细胞转换,但在捕获长时间尺度(几天到几个月)内发生的发育轨迹方面可靠性较低。
整合表观遗传学和蛋白质组学数据也可以改善轨迹的特征描述,并基于染色质可及性和蛋白质表达指导细胞转换的方向17,110。最终,具有单细胞分辨率的条形码谱系追踪提供了关于细胞转换序列的真实信息(见图3c)。
这张图展示了细胞谱系条形码(Lineage barcoding)的概念,这是一种用于追踪细胞谱系和发育轨迹的技术。

图中的每个圆圈代表一个细胞,圆圈内的彩色条形码表示该细胞的基因表达状态或特定的分子标记。
-
条形码(Barcoding):每个条形码由不同颜色的条组成,每种颜色代表一个特定的基因或分子标记。这些标记可以是细胞类型特异性的,也可以是表示细胞在发育过程中的特定状态。
-
细胞发育轨迹:箭头表示细胞从一个状态转变到另一个状态的过程。通过分析这些条形码的变化,研究人员可以推断细胞是如何从一种类型或状态转变为另一种。
-
谱系条追踪:通过比较不同细胞的条形码,可以确定哪些细胞是从同一个祖先细胞衍生出来的,从而揭示细胞的谱系关系。
整体而言,这张图强调了通过条形码技术来追踪细胞的谱系和发育轨迹,这对于理解细胞如何分化和特化以及它们在发育和疾病中的作用至关重要。
可以使用工程化基因组标签(如重组事件PolyLox111、病毒整合TraceSeq112、基于CRISPR的条形码113)或自然标记(包括体细胞突变114,115、线粒体DNA变异116和DNA甲基化模式117)实验性地追踪单细胞。
2、测量细胞可塑性
细胞可塑性指的是细胞可以独立于基因组改变而采取的潜在命运多样性。它是正常细胞对疾病反应(例如,感染中的多样化T细胞状态118)和癌细胞在肿瘤发生和治疗逃逸过程中的适应119的关键特征。已开发出多种计算方法来使用单细胞转录组学量化细胞可塑性(见表1)。
可塑性可以概念化为低维嵌入中的概率过程,其中任何给定细胞达到图中终端点的可能性可用于估计潜能(见图3d)。Palantir120、VIA121和MARGARET122是推断复杂地形、识别终端点并使用马尔可夫链模型根据每个细胞达到定义终端点的可能性为其分配概率的方法。达到每个终端状态概率相等的细胞被认为具有比概率倾斜的细胞更大的潜能。FateID93通过使用针对预定义终端状态训练的随机森林分类器来计算转换概率。GpFates将细胞命运建模为重叠高斯过程的混合,而STEMNET将命运建模为广义线性回归模型,其中系数权重代表对预定义终端状态的贡献124。
应用于单时间点收集的scRNA-seq数据的方法的一个主要缺陷是它们依赖于关于细胞转换起始和终止点的先验知识。
CellRank125,126通过利用无偏工具(如RNA速度和CytoTRACE,如下所述)的集成方法来改进这一点,以推断细胞状态的起源和目的地,然后应用马尔可夫链模型来计算命运概率。
在时间序列数据中,可以使用Waddington-OT计算命运概率,该方法将细胞转换建模为最优运输问题127。TrajectoryNet使用类似的最优运输方法,推断细胞在时间点之间的连续、非线性轨迹128。
细胞可塑性也可以作为主调节基因及其下游表达网络的函数来研究。CellOracle129利用从单细胞multi-omics分析中获得的基因调控网络知识,进行in silico转录因子扰动并展示命运改变。
2-6:测量发育潜能
发育潜能是可塑性的一种分类,它衡量细胞产生更分化细胞的能力。
能够发育成整个生物体的细胞,如受精卵,具有最高的发育潜能,而终末分化的细胞,如中性粒细胞,具有最低的发育潜能(见图3e)。
这张图展示了发育潜能的衡量标准,特别是在细胞分化过程中的潜能变化。

图中分为三个主要部分:
-
发育潜能(Developmental potential):
- 从上到下,表示细胞的发育潜能从高到低。
- Zygote(合子):具有最高的发育潜能,能发育成任何类型的细胞。
- Embryonic stem cell(胚胎干细胞):潜能稍低,能发育成多种细胞类型。
- Haematopoietic stem cell(造血干细胞):潜能进一步降低,主要分化为血液和免疫系统细胞。
- Myeloid progenitor(髓系前体):潜能更低,主要分化为髓系细胞。
- Mature neutrophil(成熟中性粒细胞):最低的发育潜能,已经高度分化,执行特定功能。
-
转录程序(Transcriptional programmes):
- 表示控制细胞命运和分化状态的转录因子。
- 包括 NANOG、SOX2、KLF4、MYC 和 OCT4 等因子,它们在细胞分化和发育过程中起着关键作用。
-
转录多样性和熵(Transcriptional diversity and entropy):
- 通过RNA和染色质(Chromatin)的可视化表示,展示了细胞在转录水平上的多样性。
- 不同颜色的波浪线表示不同类型的RNA,反映了细胞内基因表达的多样性。
- 染色质的排列表示细胞内染色体的结构,可能暗示了染色质的开放程度和可及性,这与基因表达的活跃程度相关。
整体而言,这张图强调了细胞从一种未分化状态到高度分化状态的过程中,其发育潜能是如何逐步降低的,以及这一过程中转录因子和转录多样性如何变化。这对于理解细胞如何响应发育信号和如何分化成特定类型的细胞具有重要意义。
发育潜能是干细胞和祖细胞在组织生长、稳态和再生过程中的标志130。因此,这一领域计算工具的目标是预测单个细胞在身份和功能上从最不专门化到最专门化的连续体上的分化(见表1)。
预测发育潜能的一种策略是测量早期胚胎发生过程中的活跃转录程序(见图3e)。这些工具的例子包括ORIGINS,它是由基因本体论中的“细胞分化生物学过程”衍生出的蛋白质-蛋白质网络131;mRNAsi,这是一个在多能干细胞基因表达数据上训练的逻辑回归模型132;以及PluriNet,一个由多能干细胞共享的蛋白质-蛋白质网络导出的基因集133。
然而,将这些模型应用于胚胎后发育过程假设了维持胚胎干细胞多能性的相同程序也控制成人正常细胞和肿瘤细胞的可塑性。FitDevo通过从包括胎儿和成人细胞在分化过程中的数据集训练广义线性模型,扩大了这一范围134。
单细胞分析还表明,染色质可及性和转录多样性是发育潜能的替代指标(见图3e)。直观上,基因组中表达广泛的细胞比表达多样性有限的细胞具有更高的潜能,可以转变为不同的下游细胞状态。
CytoTRACE表明,一个简单的测量——单个细胞表达的基因总数——可以概括从受精卵到终末分化细胞在各种组织和物种中的细胞发生过程135。这种转录广泛性与通过转座酶可及染色质测序(ATAC-seq)计算的全基因组染色质可及性相关。
鉴于其在寻找正常组织中较不分化细胞方面的表现,CytoTRACE被应用于人类乳腺癌数据,并识别出一群以GULP1表达为特征的肿瘤生成性腔祖细胞。转录多样性也可以通过计算香农熵来量化,香农熵是数据中不确定性的流行测量方法。这些方法在计算香农熵的数据表示上有所不同,范围从整个转录组136、最高表达基因137、或经过精心设计的基因-基因网络138、基因集-基因集网络139或蛋白质-蛋白质网络140。
这些方法的几种变体已被开发出来,以优化运行时间141和整合不同的网络分布测量方法142。一项最近的预印本研究利用带有时间点和发生学注释的公开可用数据集,构建了一个灵活的机器学习模型,该模型在单细胞RNA测序数据中学习潜在特征,并进一步提高预测发育潜能的性能143。
三、空间转录组学
细胞存在于一个由细胞外分子、结构基质和邻近细胞组成的复杂微环境中,这些因素共同塑造了细胞的表型和组织的功能特性144。
尽管单细胞RNA测序(scRNA-seq)技术不断扩展跨多种生物体和组织的细胞状态词典,但它未能捕捉到培养特定细胞身份的微环境背景。空间转录组学(ST)作为一种方法,能够同时测量目标细胞及其周围细胞的转录状态。
通过将单细胞生物学的范围从孤立细胞扩展到多细胞邻里,ST揭示了不同组织和条件下细胞重复出现且功能性的组织结构145–147。
3-1:细胞状态的空间排列特征
为了生成ST数据,已经开发出多种方法,这些方法在细胞分辨率、转录组覆盖范围、组织兼容性、成本、基础设施以及商业可用性方面存在差异。
使用荧光原位杂交(FISH)的方法,如Vizgen MERSCOPE148、NanoString CosMx149和seqFISH+150,通过结合预设计探针的连续杂交与高分辨率成像,以亚细胞分辨率捕获数百至数千个转录本。其他基于探针的方法,如STARmap151和10x Genomics Xenium152,使用原位测序作为读出。
由于这些方法目前需要创建定制的基因面板并验证基因特异性探针,因此当有先验知识可用于选择感兴趣细胞状态的标志基因时,它们最为有用。基因面板可以由特定细胞类型或感兴趣状态的已知标志物构建;或者,存在几种无聚类方法可用于选择最能捕捉可比scRNA-seq数据集中变异的基因(例如,SCMER153和geneBasis154)。
当感兴趣的细胞状态尚未定义或现有基因标志物无法区分罕见和微妙的细胞状态时,全转录组覆盖是理想的。
我们称之为“bulk ST”的一组方法,在定义的样本区域进行全转录组测序,这些区域的空间分辨率各不相同。这些方法包括激光捕获显微切割后进行批量RNA测序(LCM-seq)和基于阵列的ST平台,如10x Genomics Visium、Slide-seq V2和Stereo-seq,它们分别捕获直径为55 µm、10 µm和220 nm的点的RNA,以及分辨率为2 µm平方的10x Genomics Visium HD158。
尽管较小的点直径提高了单细胞分辨率,但这一改进被较低的转录捕获效率所抵消。与bulk ST相比,已经开发出几种方法,这些方法在组织解离后进行空间条形码和scRNA-seq。
这些方法,包括XYZeq、sc-Space和Slide-Tags,实现了高转录捕获效率,但牺牲了空间分辨率(分别为500 µm、222 µm和10 µm),并产生了解离伪迹和组织欠采样问题159–161。
不同ST技术在多样组织和条件下的性能仍有待全面评估。例如,几种ST检测已在脑部进行了广泛测试,这部分归因于现存的丰富解剖学和分子数据162,163。
然而,并非所有组织在参考图谱中都得到同等代表,且研究缺乏稳健细胞图谱注释或一致解剖结构的组织,如肿瘤组织,可能较为困难。某些组织可能还呈现更大的技术挑战,如高自荧光、寡核苷酸的不均匀扩散、降解RNA的高浓度化学物质,或由于材料属性(例如,骨骼)难以切片的结构。
在设计ST实验时还有许多其他考虑因素,感兴趣的老师/同学可以参考其他关于此主题的评论164–166。
3-2:克服技术障碍的计算工具
为解决空间转录组学(ST)技术的固有局限性,包括细胞分辨率、转录本覆盖范围以及独立样本的对齐问题,已开发出多种计算方法(见表1)。
例如,一种常见策略是使用与生物和实验特征相匹配的参考单细胞RNA测序(scRNA-seq)图谱来提高ST的分辨率或转录本覆盖范围。尽管标准的批量RNA测序去卷积工具可以应用于批量ST167,168,但已开发出专门的工具,这些工具利用scRNA-seq数据来解析空间点169,无论是到特定细胞类型的分数(例如,cell2location170, RCTD171),单个单细胞转录组(例如,CytoSPACE172, Tangram173和CellTrek174),亚点或像素级别的表达(例如,BayesSpace175, XFuse176, TESLA177和iStar178),或是推断的特定细胞类型的表达轮廓(ST deconvolve179)。
在发育生物学180,181、肠道生物学182,183和肿瘤生物学184–189等领域,已经应用批量ST去卷积方法来定位感兴趣的细胞状态。最近的研究还表明,利用深度学习方法将组织学与ST数据整合可以增强ST去卷积工具的空间和表型分辨率176–178。
对于通常以单细胞分辨率测量转录组子集的基于探针的ST,存在多种方法用于估算未测量基因的表达172,173,190,191。此外,最近的基准测试表明,通过将scRNA-seq与ST数据整合,然后平均计算ST细胞的最近scRNA-seq邻居的基因表达,单细胞RNA测序的批次校正技术可以实现具有竞争力的性能169,191,192。
另一个关键的计算挑战是构建多个2D ST样本的连贯空间图谱,这些样本来自相邻组织或跨越时间序列。可以根据注册的模态、组织切片的变形程度或个体间组织结构的变化来选择适当的ST对齐方法193–196。对齐方法还可以用于将2D ST数据整合到共同的坐标框架中,甚至可以用于(近似)对齐不同生物体相似解剖区域的2D切片。
3-3:通过比较多细胞邻里发现重复的空间组织
将单细胞研究扩展到相互作用细胞的多细胞枢纽,正在提高我们对各种生物机制的理解144,包括细胞可塑性197、命运决定198和肿瘤免疫学199。尽管组织学结构为ST分析提供了一个起点,但它们可能无法实现对细胞微环境如何决定其表型的详细分析。
一个多细胞邻里,由一个细胞及其局部微环境组成,是ST数据分析的基本单位。多细胞邻里的定义应以数据驱动的方式考虑技术研究的尺度和现象(见图4a)。
这张图展示了从单细胞空间转录组学(single-cell spatial transcriptomics, ST)数据中识别空间生态型(spatial ecotypes)的分析流程。

流程分为三个主要步骤:
-
定义多细胞邻域(Define multicellular neighbourhoods):
- 生物信息学工作流(Bioinformatics workflow):通过空间最近邻域图(spatial nearest-neighbour graph)来定义多细胞邻域。
- 深度学习工作流(Deep learning-enabled workflow):使用卷积训练层(convolutional trained layer)和非线性变换(nonlinearity)来识别多细胞邻域。
-
识别邻域特征(Identify neighbourhood features of interest):
- 组成(Composition):确定主要细胞类型(如B细胞、T细胞、成纤维细胞等)。
- 细胞状态(Cell states):识别转录亚型亚群,反映细胞的特定功能或状态。
-
识别空间生态型(Identify spatial ecotypes):
- 通过社区检测(Community detection)和推断(Inference)识别空间生态型,即多细胞邻域的集群。
整体而言,这张图强调了如何结合生物信息学方法和深度学习技术来分析单细胞空间转录组学数据,以识别和理解细胞在组织中的分布和相互作用。这对于揭示组织结构、细胞间通讯和疾病机制具有重要意义。
在批量ST数据的背景下,一个多细胞邻里可以定义为阵列中的单个点,一个“超点”(一个点及其最近邻居)147,175,200,或一个或多个空间点的去卷积结果。
对于单细胞分辨率的ST数据,许多多细胞邻里的定义使用固定的半径(50–200 µm)201,202、固定数量的邻近细胞(10–200个最近邻居)149,203,204在2D或3D空间中,或由Delaunay三角测量定义的邻里203。
选择用于表征多细胞邻里的适当特征集是一个依赖于数据和应用程序的过程(见图4a)。对于批量ST,如果每个点被视为一个多细胞邻里,那么点的基因表达值是进行下游分析(如聚类)的自然特征186。
对于单细胞分辨率的ST数据,一个多细胞邻里可以由其组成表示,即预先选择的细胞类型和状态的丰度,这些类型和状态使用已知标志物定义202。这种方法在空间蛋白质组学中很常见,具有识别组织学上不同区域202、显示不同细胞间信号模式的不同区域206、捕获不同淋巴器官之间组织结构的相似性和差异性207,以及识别与回顾性癌症队列中治疗结果相关的多细胞邻里主题203,205的表达能力。
其他策略将细胞的基因表达值聚合起来代表多细胞邻里,使用加权平均102,208、基因-基因协方差矩阵190或深度学习209–211(见图4a中的“深度学习启用的工作流程”)。
使用基因表达值分析多细胞邻里可以自动识别参考数据集中不存在的细胞类型或依赖于上下文的转录状态74。此外,基因表达表征适合于空间数据中的细胞-细胞相互作用分析212,213。包含基因表达状态的表征可以编码比细胞类型频率更多的复杂性,使研究人员能够区分具有相似细胞类型组成的细胞社区。
例如,可以通过同时考虑细胞的转录状态、细胞组成和细胞在结构中的空间组织准确确定三级淋巴结构的成熟状态(见图4b,c)。

图b展示了肿瘤切片的细胞类型注释(左侧)和空间生态型注释(右侧)。图c描述了来自肿瘤内或周围基质中的四个不同三级淋巴结构中的多细胞邻域。重要的是,多细胞邻域1和2在主要细胞类型的组成上相似,但它们的详细结构组织和细胞状态不同;类似的区分也适用于多细胞邻域3和4。
因此,空间生态型(相似多细胞邻域的群体)是由细胞类型组成、细胞状态异质性和空间组织共同定义的。B代表B细胞;DC代表树突细胞;Epi.代表上皮细胞;Fib.代表成纤维细胞;T代表T细胞;ST代表空间转录组学。
表达特征的潜在缺点是存在批次效应,其中多细胞邻里主要按技术协变量(例如,样本)而不是生物学特征分离。在深度学习分析中,这种效果已在训练模型的过拟合中观察到205。
存在多种策略来减轻这种设置中的批次效应,例如在多细胞邻里分析之前应用数据整合工具197,211,以及在一个最近的预印本研究中展示的,使用深度学习同时嵌入多细胞邻里和执行数据整合的新兴策略215。
在创建了多细胞邻里的表征之后,一个重要的目标是识别跨空间域、样本或个体中反复出现的细胞状态社区(见图4a)。我们将由相关细胞状态特征的定义为“空间生态型”。这一概念借鉴了生态学中“生态型”作为空间依赖的物种亚群的概念216,217,以及引入“生态型”一词用于肿瘤微环境中相关转录状态的工作218–221。
转录生态型和空间生态型的定义也与“多细胞程序”222相吻合,后者是在多个样本或空间区域内不同细胞类型上共同相关的转录状态集合。这些研究和其他研究223,224的基本见解是,可以通过确定在许多独立样本或空间区域内频率相关的细胞状态来识别相关的细胞状态。
空间生态型和相关的多细胞邻里概念在癌症研究中特别有用,其中它们可以应用于在缺乏良好定义的组织学特征的情况下识别反复出现的表型。例如,在一组皮肤鳞状细胞癌患者中,一种肿瘤特异性的角蛋白细胞状态在多个样本的肿瘤-基质界面处的多细胞邻里中定位225。在一项跨越多个乳腺癌样本的多细胞枢纽整合研究中,一种空间生态型在化生性乳腺癌中富集了FGF2+调节性T(Treg)细胞,沿着FGF2受体的梯度排列,这表明Treg细胞浸润参与在这一背景下构建肿瘤微环境226。
综上所述,计算工具和方法的进步正在帮助我们克服空间转录组学技术的基本限制,并使我们能够从空间数据中提取更丰富的生物学信息。通过定义和分析多细胞邻里,我们能够更好地理解细胞之间的相互作用以及它们在健康和疾病状态下的空间组织。这些发现为未来的研究提供了新的方向,旨在揭示生物系统中细胞行为的复杂网络。
3-4:空间表征表型转换
当细胞经历表型转换时,识别其多细胞邻里在组成和基因表达方面的相应变化,将为理解动态细胞过程的机制提供新的见解。
生物学实例中出现的这种挑战是多样的,包括干细胞向祖细胞和后代细胞的分化,免疫细胞对刺激的反应激活,以及癌细胞的克隆进化。这些分析属于空间轨迹分析的范畴(见表1)。
来自单细胞RNA测序(scRNA-seq)数据的轨迹分析可以通过应用空间转录组(ST)去卷积或批次整合方法188,227转移到ST中的相应细胞;轨迹分析也可以不使用空间信息直接在ST数据上执行202,227,尽管当前ST检测的技术限制(低转录捕获或缺乏单细胞分辨率)可能会影响结果的准确性。
最近针对ST数据的轨迹分析建立在非空间方法的基础上,通过使用各种技术策略来促进“空间连贯性”(即空间上接近的细胞在轨迹中具有相似的位置)210,212,228。然而,无论是在单个样本内还是在独立样本之间,模拟多细胞邻里的表型轨迹仍然是一个挑战。
最后,当存在基因组标记,如线粒体DNA变异、拷贝数改变和遗传突变时,可以使用这些标记在空间数据中追踪克隆进化229–233。将详细的系统发育与ST数据相结合,将有助于进一步研究克隆进化对表型转变的影响程度234。
四、将单细胞和空间转录组学转化为临床应用
迄今为止,单细胞RNA测序(scRNA-seq)已应用于超过103种人类疾病的研究51,并被纳入超过58项血液学、肿瘤学和免疫学的临床试验235。
从患者样本中构建单细胞分辨率的大型细胞图谱,有望推动向精准医学的转变,其中特定细胞和转录本的存在和比例将指导疾病分类、治疗和预后。单细胞RNA测序已在阐明疾病进展和传播机制、识别与不良预后相关的稀有或微妙细胞状态以及揭示潜在治疗脆弱性方面得到应用236。
例如,肿瘤微环境的单细胞剖析已在多种癌症类型中揭示了对临床相关的免疫224,237,238和非免疫239,240表型;它还使得静止和治疗抵抗性癌细胞干细胞241,242以及具有预后和治疗靶点特征的循环肿瘤细胞的表征成为可能。
单细胞转录组学还促进了正常人类神经和骨骼干细胞及祖细胞的描述88,89,并为将干细胞分化为期望的细胞命运,最终用于细胞移植应用提供了高分辨率的路线图244。
尽管最初是在小队列中发现,但scRNA-seq的发现可以通过“数字细胞测量”(表1,图5,方框1)从大型临床数据库中的批量RNA混合物中解卷积细胞组成,进行外部验证和大规模测量。确实,scRNA-seq在临床的直接应用受到成本、标准化和组织解离人工产物等因素的阻碍(图5a)。

这张图展示了从发现(Discovery)到验证(Validation)再到应用(Applications)的科研流程,特别是在生物标志物的选择和应用方面。
发现(Discovery)阶段:
- 使用空间转录组学(Spatial transcriptomics)和单细胞RNA测序(scRNA-seq)技术来识别新的生物标志物。
- 这一阶段的目标是发现空间生态型(Spatial ecotypes)、细胞状态(Cell states)和细胞类型(Cell types)。
- 通常需要较高的细胞分辨率、每个样本的成本较高,以及较多的样本数量。
验证(Validation)阶段:
- 通过多重免疫标记(Multiplex immunolabelling)、定量PCR(qPCR)和批量基因组学(Bulk genomics)等技术来验证发现的生物标志物。
- 这一阶段的目标是确认这些标志物的可靠性和有效性,通常需要较低的每个样本成本和较少的样本数量。
应用(Applications)阶段:
- 将验证后的生物标志物应用于研究(Research)和临床(Clinical)领域。
- 在研究领域,用于提出机制假设(Mechanistic hypotheses)和发现新的药物靶点(New drug targets)。
- 在临床领域,用于开发预测性和预后标志物(Predictive and prognostic markers)以及疾病监测(Disease monitoring)。
整体而言,这张图强调了生物标志物从发现到验证再到应用的转化过程,这是生物医学研究中将基础科学发现转化为实际应用的重要途径。
然而,scRNA-seq可以优先考虑使用临床环境中已经可用的更简单检测方法评估的转录本、细胞状态和蛋白质,如免疫组化、FISH、流式细胞术和多基因组合测序。
空间转录组学(ST)在发现临床相关的空间定义细胞状态和生态型方面发挥着类似的作用,这些状态和类型可以通过更简单的检测方法进行总结,以指导临床决策(图5b,方框1)。

这张图展示了如何利用单细胞RNA测序(scRNA-seq)数据来分析临床队列中细胞状态与患者预后的关系。
-
临床队列(Clinical cohort):从具有混合结果的临床队列中收集样本。
-
单细胞RNA测序(scRNA-seq):对样本进行单细胞RNA测序,得到每个细胞的基因表达数据。
-
结果相关细胞状态(Outcome-associated cell states):使用降维技术(如UMAP)对单细胞数据进行分析,识别与临床结果相关联的细胞状态。
-
细胞状态参考图谱(Cell state reference profiles):建立细胞状态的参考图谱,用于后续比较和分析。
-
大量临床队列(Large clinical cohort):在更大的临床队列中应用这些细胞状态参考图谱。
-
通过去卷积的细胞状态丰度(Cell state abundance by deconvolution):使用去卷积方法从大量样本的基因组学数据中估算细胞状态的丰度。
-
结果关联(Outcome association):分析细胞状态丰度与患者生存率等临床结果之间的关联。
整体而言,这张图强调了单细胞RNA测序技术在解析细胞状态和它们与临床结果之间关系中的应用,以及如何将这些信息应用于更大的临床队列中,以揭示细胞状态与疾病预后之间的关系。
空间生物标志物已在包括脑245、乳腺246、结直肠247和肺癌197,248在内的几种癌症中显示出与临床结果和治疗反应的相关性。两种空间生态型的基因表达特征,分别与口腔鳞状细胞癌的肿瘤边界和非小细胞肺癌的上皮-间质转化相关,在大规模保留的批量RNA-seq队列中预测了生存期188,202。
在结直肠癌中,PD-1+ T细胞和PD-L1+髓系细胞之间的免疫相互作用的空间模式为免疫逃逸提供了洞察249。scRNA-seq和ST在石蜡包埋组织中的应用技术进步,使得对存放在生物样本库中的临床样本进行回顾性分析成为可能。
人类蛋白质图谱等大型工作正在创建一个平台,通过整合蛋白质、转录和地理空间特征以及生物学和临床注释,用于生物标志物发现250。
五、人工智能与单细胞转录组学的未来
随着单细胞RNA测序(scRNA-seq)和空间转录组学(ST)数据集在深度、规模、分辨率和复杂性方面的持续增长(例如,通过引入多组学能力2),人工智能(AI)的并行飞跃正宣告着机器辅助单细胞生物学新纪元的到来(图6a)。

这张图展示了从4D组织图谱到功能假设验证的整个研究流程,特别是在单细胞空间多组学和人工智能/机器学习(AI/ML)在细胞状态发现中的应用。
-
4D组织图谱(4D tissue atlas):
- 通过3D空间采样和时间采样(Temporal sampling)获取组织的详细结构信息。
-
单细胞空间多组学(Single-cell spatial multi-omics):
- 利用单细胞技术从形态学(Morphology)、基因组(Genome)、表观基因组(Epigenome)、转录组(Transcriptome)和蛋白质组(Proteome)等多个层面分析细胞。
-
AI/ML发现细胞状态(AI/ML discovery of cell states):
- 应用降维技术(如UMAP)对单细胞数据进行分析,识别不同的细胞状态。
-
功能假设验证(Functional hypotheses for validation):
- 基于AI/ML分析结果提出的功能假设,通过实验进行验证。
整体而言,这张图强调了单细胞空间多组学技术在解析组织结构和细胞状态中的作用,以及AI/ML在发现细胞状态和生成可验证功能假设中的应用。这对于理解组织功能、疾病机制和开发新的治疗方法具有重要意义。
深度学习在单细胞转录组学中的兴起得益于数据可用性的增加、新模型架构(例如,Transformer251和视觉Transformer178)的发展,以及使非专家能够训练、应用和解释深度学习模型的软件252。
深度学习可以应用于各种问题,取决于模型架构(例如,卷积神经网络和图神经网络)或训练框架(监督或无监督)。例如,监督方法可以从微妙的分子或空间模式预测治疗结果的差异205,253,而无监督方法可以在不需要手动批次校正的情况下,揭示跨多样本数据集的共同生物学特征254。
深度学习模型的性能对“超参数”的选择(包括模型的深度和维度以及训练过程的精确细节)高度敏感。因此,由训练模型组成的方法具有优势,因为它们消除了或减少了对进一步超参数调整的需求。
基础模型在单细胞转录组学研究社区及更广泛的领域引起了极大的兴趣。
基础模型是在大量无标签数据上训练的深度学习模型,旨在为下游应用形成“基础”,可以不经任何额外训练直接应用或使用少量标签数据“微调”模型以适应特定任务255。单细胞转录组学的基础模型通常是在多个数据集上训练的251,254,256–262。
在单细胞转录组学分析中使用基础模型的一个假设是,这些模型能够自动学习将来自不同平台、组织和条件的scRNA-seq数据组织成一个连贯的嵌入(图6b和表1)。
这张图展示了如何使用单细胞图谱(single-cell atlases)和其他数据来训练基础模型(foundation model),并应用于转录状态空间(Transcriptional state space)的分析。

训练数据(Training data):
- 单细胞图谱(Single-cell atlases):这些是经过系统采样和人工注释的,提供了高质量的参考数据集。
- 其他数据(Other data):可能包括采样和注释不一致的数据,这些数据用于增加模型的泛化能力。
基础模型(Foundation model):
- 使用训练数据通过降维技术(如UMAP)进行处理,然后输入到基础模型中进行训练。
- 基础模型学习数据中的转录模式,能够捕捉细胞状态的主要特征。
转录状态空间(Transcriptional state space):
- 训练好的基础模型可以用于推断转录状态空间,这是一个高维空间,其中每个点代表一个细胞的转录状态。
- 通过模型,可以探索扰动(Perturbations)、疾病(Disease)、微环境(Microenvironment)和谱系(Lineage)等因素如何影响细胞的转录状态。
整体而言,这张图强调了利用高质量的单细胞参考数据集来训练深度学习模型,进而分析和推断细胞在不同生物学条件下的转录状态,这对于理解细胞如何响应不同的生物学和临床条件具有重要意义。
在基础模型嵌入中,研究人员可以搜索其他数据集中与输入细胞相似的细胞254,执行跨数据集的数据整合257,262,将标签(如细胞类型或状态)从标记数据集转移到未标记数据集262–264,或预测对细胞转录状态的影响251,258(图6c和表1)。
这张图展示了如何利用单细胞图谱和空间转录组学数据进行数据分析和实验模拟的流程。

查询公共数据集(Query public datasets):
- 通过UMAP(Uniform Manifold Approximation and Projection)可视化技术展示不同数据集(如GEO 001, GEO 041等)的单细胞数据。
- 目标是找到与特定细胞状态匹配的细胞。
分析新数据(Analyze new data):
- 将新的数据集与人类(或其他器官)图谱进行整合。
- 使用图谱的注释来标注细胞类型。
模拟实验(Simulate experiments):
- 推断给定细胞周围最可能的多细胞邻域。
- 探讨特定细胞(如耗竭的T细胞)是否会被特定途径的抑制剂重新激活。
整体而言,这张图强调了单细胞图谱数据在理解细胞状态、进行数据集比较和整合以及在没有直接实验条件的情况下模拟生物学实验结果中的应用。这对于生物医学研究中的数据解读和实验设计具有重要意义。
另一个假设是,在大量无标签数据上预训练模型可以提高其从少量标签数据中学习的能力263(尽管并非所有模型都能实现这一点265)。
正如最近的一篇预印本所提出的,这些嵌入空间捕捉生物学信号的程度仍然是一个开放的问题266,并且这些嵌入空间对训练数据中某些组织或疾病状态的过度表示的敏感性尚未被系统性地解决。嵌入模型可能无法捕捉训练数据中代表性不足的细胞或用于微调的数据中的细微生物学,而对于生成模型,一个额外的担忧是它们产生没有现实世界对应物的结果的倾向。
正在进行的研究265–267旨在重现已发表(或预印本)的基础模型,将它们与黄金标准进行基准测试,并实施护栏以防止或至少识别潜在的幻觉,这对于评估它们的能力以及在各种下游应用中推广到新数据集至关重要。
ST基础模型的发展受到相对缺乏组织化、大规模图谱的复杂化。
最近的一篇预印本报告了在scRNA-seq和ST数据上训练单细胞基础模型的工作268。这项工作表明,基础模型嵌入编码了单细胞微环境背景的信息;类似的模型可以训练来明确纳入ST数据中包含的微环境背景和scRNA-seq的转录数据(例如,通过使用多种技术特定的解码器190),这可能导致能够为scRNA-seq数据模拟多细胞邻里的空间感知基础模型。
与scRNA-seq一样,ST基础模型可能会受到不平衡训练数据的偏见;ST数据的额外挑战是探针基础的ST的普遍性,其面板偏向于特定基因。这可能通过关注当前ST面板中过度代表的基因途径,而掩盖了空间调节转录状态的广泛规则。
此外,空间解析扰动模型可以视为现有单细胞扰动模型的延伸269。这类模型将能够进行微环境扰动的in silico测试,包括组合扰动。形成微环境决定因素对依赖背景的细胞状态的因果假设的能力,可能会彻底改变我们对从干细胞生态位到免疫系统的功能和功能障碍的细胞-细胞相互作用的理解。
尽管深度学习模型通常能够具备优越的性能,但理解驱动模型预测的因素,包括细胞状态、基因集、空间生态型、原始文献、数据库或规则(例如,组合和互斥性)仍然是一个挑战。这些问题属于模型可解释性的范畴,这是机器学习社区日益活跃的研究领域270。可解释性不仅对于揭示新的生物学至关重要,而且对于在临床环境中建立信心和信任同样至关重要271。
六、结论
单细胞转录组学和空间转录组学技术正在革新我们对复杂组织中细胞状态的起源、调控和维护的理解。通过精心的实验设计和计算方法的策略性利用,研究人员能够充分发挥单细胞RNA测序(scRNA-seq)和空间转录组(ST)数据的作用,揭示在各种背景下关键的细胞状态和多细胞群落。
尽管数据分析存在固有的挑战,但迅速发展的实验和计算工具,以及人类细胞图谱、人类蛋白质图谱和人类肿瘤图谱网络等大型合作项目,正在促进将这些细胞状态置于人类健康和疾病更广泛谱系中的情境化。随着来自不同组织、物种和条件下单细胞的大体积基因组、空间和时间数据的整合,我们正开始揭开细胞之谜。